Prawidłowa odpowiedź: Jest najczęstszym zespołem genetycznym związanym z rakiem jelita grubego
Komentarz
W ok. 5% przypadków rak jelita grubego rozwija się na podłożu dziedzicznych zespołów genetycznych. Najczęstszym z nich jest zespół Lyncha, nazywany również dziedzicznym rakiem jelita grubego niezwiązanym z polipowatością (hereditary non-polyposis colorectal cancer – HNPCC). Nieco rzadszym zespołem genetycznym, odpowiadającym za niespełna 1% raków jelita grubego, jest zespół rodzinnej polipowatości gruczolakowatej (familial adenomatous polyposis syndrome – FAP).
Zespół FAP charakteryzuje się rozwojem setek, a niekiedy tysięcy polipów gruczolakowatych w jelicie grubym. Pozostawienie choroby nieleczonej związane jest z blisko 100% ryzykiem rozwoju raka jelita grubego w życiu dorosłym (przeważnie w 40.–50. roku życia). Z uwagi na to ryzyko, postępowaniem z wyboru jest profilaktyczne usunięcie okrężnicy (z pozostawieniem odbytnicy – kolektomia) lub całego jelita grubego (proktokolektomia) przeważnie w 2.–3. dekadzie życia. U pacjentów z pozostawioną odbytnicą (lub wytworzonym zbiornikiem jelitowym typu pouch) powinno się wykonywać corocznie rektoskopię z usuwaniem kolejnych polipów na tym odcinku.
Polipy rozwijają się również w obrębie dwunastnicy i żołądka. Z tego powodu pacjenci ci wymagają regularnej kontroli gastroskopowej (pierwsze badanie w 25–30 roku życia). W dwunastnicy polipy przeważnie przybierają postać rozproszonych gruczolaków (także brodawki Vatera) o różnym stopniu zaawansowania (ryc. 1) i związane są z ok. 5% ryzykiem rozwoju raka dwunastnicy, natomiast w żołądku występują pod postacią licznych polipów z gruczołów dna żołądka (fundic gland polyps; ryc. 2), których ryzyko transformacji nowotworowej szczęśliwie jest małe. Do obciążeń spoza przewodu pokarmowego w zespole FAP należą m.in. łagodne zmiany, jak przerost nabłonka barwnikowego siatkówki oka (70–80%) czy guzy desmoidalne (10–15%), ale również inne nowotwory złośliwe, jak rak tarczycy (2–3%) czy guzy mózgu (<1%). Z powodu tego pierwszego nowotworu zaleca się dodatkowo wykonywanie USG tarczycy każdego roku.
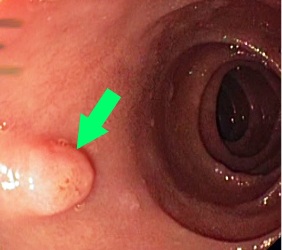 Ryc. 1. Zespół FAP – polip gruczolakowaty dwunastnicy
Ryc. 1. Zespół FAP – polip gruczolakowaty dwunastnicy
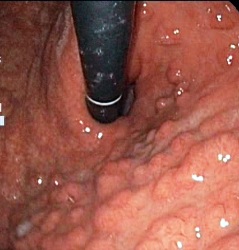 Ryc. 2. Zespół FAP – polipy dna żołądka
Ryc. 2. Zespół FAP – polipy dna żołądka
Typowa postać zespołu FAP dziedziczona jest na drodze autosomalnej dominującej i spowodowana jest mutacją w obrębie genu APC. Do innych postaci należy łagodniejsza tzw. atenuowana postać FAP – AFAP oraz dziedziczona w sposób autosomalny recesywny polipowatość związana z mutacją genu MUTYH – zespół MAP.
Należy pamiętać, że rozpoznanie zespołu FAP u pacjenta obliguje do prowadzenia nadzoru endoskopowego także wśród najbliższych członków jego rodziny (krewnych I stopnia). Badaniem z wyboru jest sigmoidoskopia wykonywana co 1–2 lata, zaczynając od dzieci w 10.–12. roku życia. Stwierdzenie polipów gruczolakowatych powinno skutkować wykonaniem pełnej kolonoskopii i planowaniem kolektomii w przyszłości. U dorosłych członków rodziny kolonoskopia może być badaniem pierwszego wyboru.
Piśmiennictwo:
1. Syngal S., Brand R.E., Church J.M. i wsp.: ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am. J. Gastroenterol. 2015; 110 (2): 223–262.
2. Vasen H.F., Möslein G., Alonso A. i wsp.: Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut 2008; 57 (5): 704–713.





