
Przyczyną choroby Pompego jest niedobór kwaśnej alfa-glukozydazy, hydrolazy lizosomalnej, która uczestniczy w procesie rozkładu glikogenu do glukozy. W efekcie glikogen odkłada się w nadmiarze w komórkach różnych tkanek, zwłaszcza mięśni szkieletowych, w tym mięśni oddechowych, i w mięśniu sercowym (ryc. 1A), powodując przerost i niewydolność serca (w przypadku skrajnego niedoboru), niedowład wiotki oraz niewydolność oddechową.
Enzymatyczna terapia zastępcza polega na dostarczeniu syntetycznego analogu brakującego enzymu drogą dożylną. Cząsteczki enzymu zawierają reszty mannozo-6-fosforanu, które są rozpoznawane przez odpowiednie receptory na powierzchni komórek, dzięki czemu syntetyczny enzym przenoszony jest bezpośrednio do lizosomów we wnętrzu komórki (ryc. 1B).
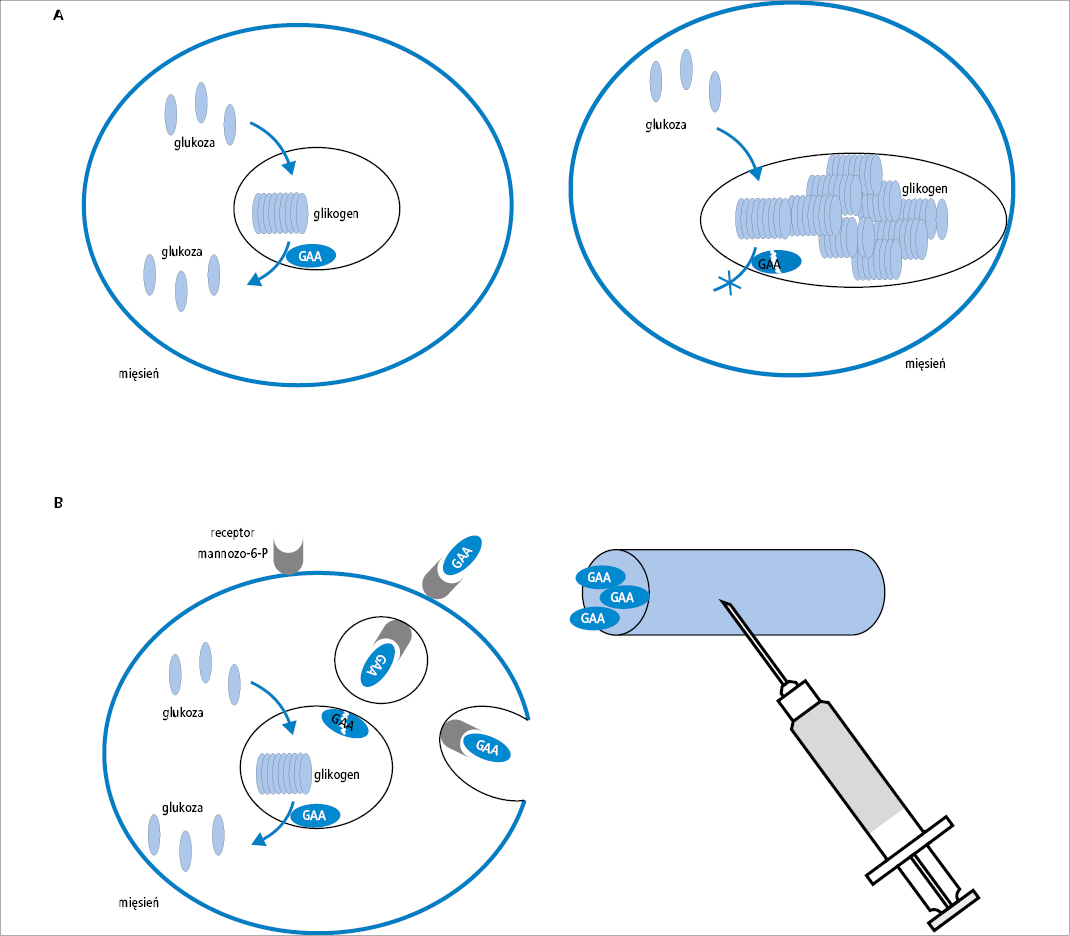
Ryc. 1. Patomechanizm choroby Pompego i zasada działania enzymatycznej terapii zastępczej. W następstwie wrodzonego niedoboru alfa-glukozydazy (GAA) w lizosomach mięśni szkieletowych i mięśnia sercowego gromadzi się w nadmiarze glikogen (A). Leczenie polega na okresowym podawaniu syntetycznego analogu kwaśnej alfa-glukozydazy, który za pośrednictwem receptorów mannozo-6-fosforanu przenoszony jest do lizosomów, kompensując wrodzony defekt genetyczny (B)















