Genetic muscle disorder mimicking atrial arrhythmias with conduction defects requiring pacemaker implantation



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Genetic muscle disorder mimicking atrial arrhythmias with conduction defects requiring pacemaker implantation
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia, which is associated with cardiovascular comorbidities, and its prevalence increases with age.1 We present the case of a young 28‑year‑old man who was admitted due to newly diagnosed AF following pre‑employment medical examination. He had no history of drug intake. He only complained of occasional dizziness and fatigue during vigorous activities. Upon further questioning, a history of weight loss despite increased food consumption in the last 6 months was elicited. Clinical examination revealed elevated blood pressure of 140/90 mm Hg, bilateral elbow and ankle contractures with asymmetric muscle wasting, especially of the right arm and left lower limb (Figure 1A and 1B). In hindsight, the patient recalled this abnormality since his late teens, but no further investigations were done at that time. Family history was relevant with cases of a sudden unexplained death of male family members on his mother’s side before the age of 40 years. At the initial referral visit, the electrocardiogram demonstrated a regular junctional escape rhythm of 35 bpm and low amplitude baseline deflections suggestive of atrial flutter (Figure 1C). Holter 12‑lead electrocardiogram monitoring confirmed episodes of AF, atrial flutter, atrial tachycardia with a slow ventricular response and significant pauses of up to 3.86 s. Doppler echocardiography revealed a normal left ventricular ejection fraction of 63%, marked biatrial enlargement, and complete absence of A waves that indicated loss of right and left atrial mechanical activity. Late gadolinium-enhanced cardiac magnetic resonance showed no signs of myocardial fibrosis or inflammation. Serum creatine kinase concentration was elevated at 724 U/l (reference range, 39–308 U/l) and high‑sensitivity troponin T concentration was high at 90.32 ng/l (reference range <14 ng/l).
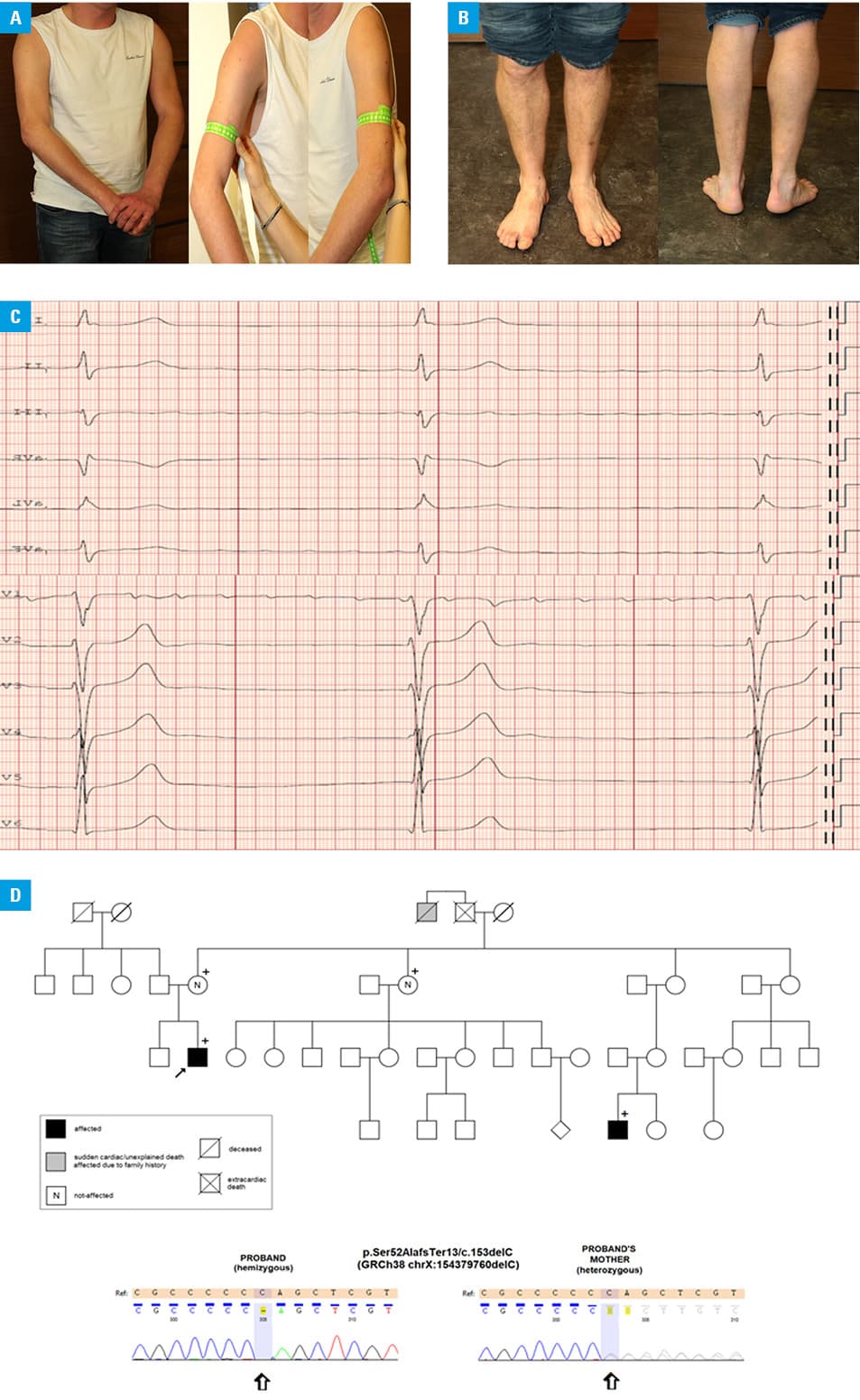
The constellation of the family and personal history as well as findings of physical examination and imaging findings suggested a diagnosis of Emery–Dreifuss muscular dystrophy (EDMD). Following genetic counseling, the patient underwent testing of the EDMD gene, which encodes the muscle‑specific emerin protein. Sanger sequencing revealed the hemizygous p.Ser52AlafsTer13/c.153delC mutation (Figure 1D), resulting in a premature truncation of the protein.2
As atrial electrical silence is frequently observed in EDMD, ablation could not be a therapeutic option for our patient. The patient was referred for pacemaker implantation. Once the right ventricular lead had been placed, electrical cardioversion of AF restored slow sinus rhythm temporarily and the patient had a permanent dual‑chamber pacemaker implanted. Anticoagulation therapy was administered to prevent thromboembolic events, based on the assessment of stroke risk in patients with EDMD, the presence of hypertension, and the patient’s choice.3,4
Genetic tests performed in the mother, the mother’s sister, and a male cousin showed a hemizygous mutation suggesting the diagnosis of EDMD in the cousin and revealed that both women were heterozygous carriers (Figure 1D). On family assessment, the patient’s male cousin, aged 20, revealed joint contractures, mild limb muscle atrophy, and bradyarrhythmias, and both female relatives were asymptomatic.
Emery–Dreifuss muscular dystrophy is a condition characterized by the clinical triad of early‑onset contractures, progressive weakness and wasting in humeroperoneal muscles, and cardiac disease with conduction defects, arrhythmia and cardiomyopathy.2 The disease has X‑linked recessive inheritance pattern and thus typically affects men (cardiac disease may manifest in some female carrier as well).
Due to the specific clinical manifestation (including muscle atrophy), EDMD could be categorized into group 2 (arrhythmias in specific clinical settings) and subgroup O (other) according to the classification of rare cardiovascular diseases and disorders (RCDD classification, VI‑2O).5
Our case demonstrates that atrial arrhythmias may be the first clinical manifestation of EDMD. Appropriate clinical assessment together with taking into account a family history, serum levels of muscle‑specific biomarkers and genetic testing may help to establish the primary cause of the disease.
- Kirchhof P, Benussi S, Kotecha D, et al. ESC Scientific Document Group. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. 2016; 37: 2893‑2962. | Crossref
- Manilal S, Recan D, Sewry CA, et al. Mutations in Emery–Dreifuss muscular dystrophy and their effects on emerin protein expression. Hum Mol Genet. 1998; 7: 855‑864. | Crossref
- Boriani G, Gallina M, Merlin L, et al. Clinical relevance of atrial fibrillation/flutter, stroke, pacemaker implant, and heart failure in Emery–Dreifuss muscular dystrophy: a long‑term longitudinal study. Stroke. 2003; 34: 901‑908. | Crossref
- Feingold B, Mahle WT, Auerbach S, et al. Management of cardiac involvement associated with neuromuscular diseases: a scientific statement from the American Heart Association. Circulation. 2017; 136: e200‑e231. | Crossref
- Podolec P, Baranchuk A, Brugada J, et al. Clinical classification of rare cardiac arrhythmogenic and conduction disorders, and rare arrhythmias. Pol Arch Intern Med. 2019; 129: 154‑159. | Crossref
ARTICLE INFORMATION