A 21‑year‑old, nonsmoking, and previously healthy woman was admitted to our hospital due to a dry cough and enlarged hilar and mediastinal lymph nodes on chest X‑ray (Figure 1A).
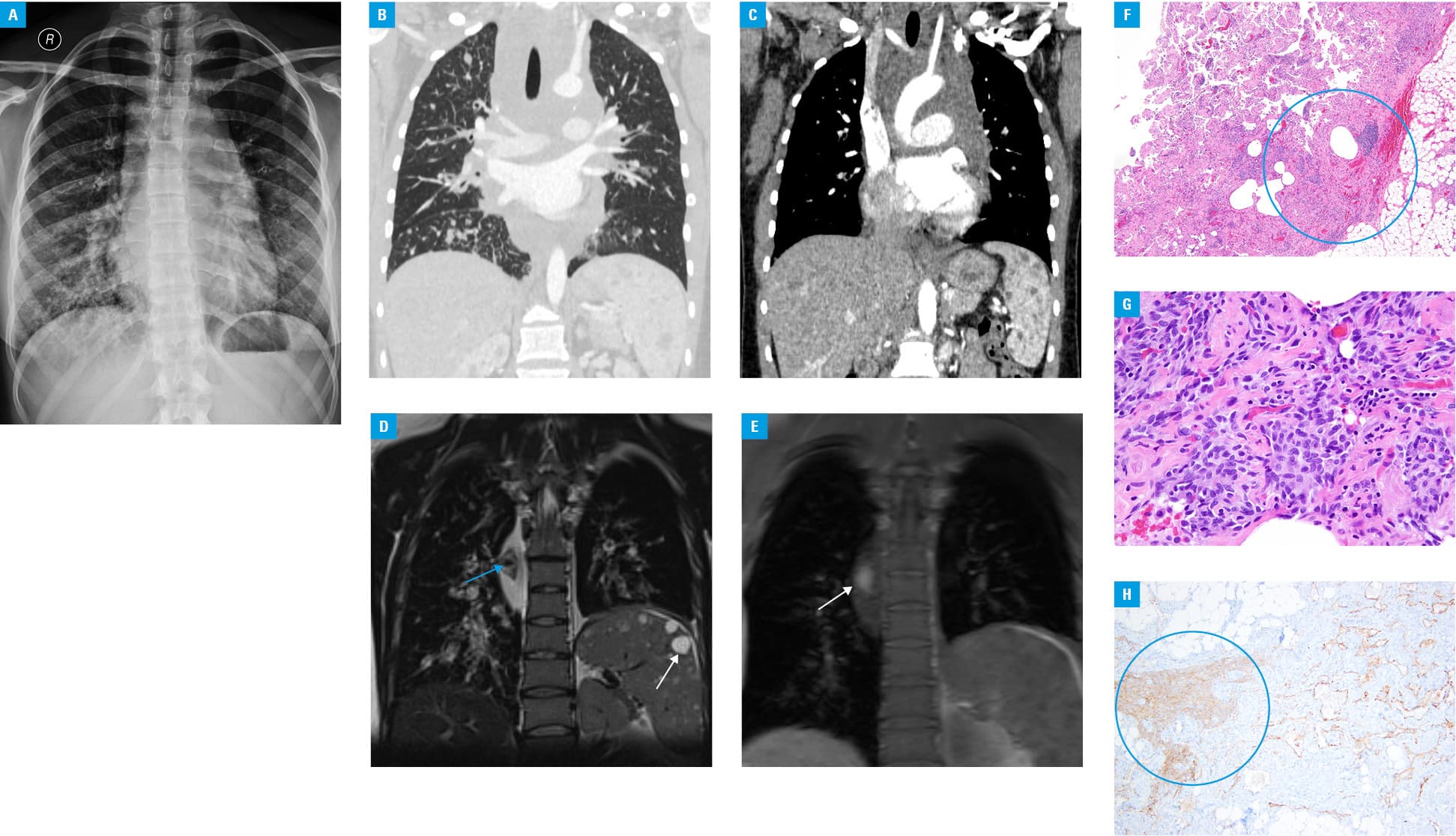
Laboratory tests revealed an elevated D‑dimer level (14905 ng/ml; reference value <500 ng/ml) and a low fibrinogen concentration (1.61 g/l; reference range, 2.2–4.9 g/l). Chest computed tomography detected interlobar and peribronchovascular thickening (predominantly in the right lung), a large mediastinal mass, and multiple hypodense splenic lesions (Figure 1B and 1C). Magnetic resonance imaging (MRI) showed a large heterogeneous mediastinal mass with diffuse bronchial wall thickening (Figure 1D). In addition, fat saturation imaging disclosed a high‑level signal from within the mediastinal mass attributable to methemoglobin, and multiple splenic lesions (Figure 1E). Histological examination of the samples obtained via mediastinoscopy revealed irregular, thin‑walled vascular spaces covered by spindle‑shaped lymphatic endothelial cells that were focally clustered (Figure 1F and 1G) and that were immunostained for CD31, podoplanin (D2‑40) (Figure 1H), and prospero homeobox protein 1 (PROX1). The histological and immunohistochemical data together with characteristic MRI findings included a heterogeneous mediastinal mass containing methemoglobin, multiple splenic lesions, an elevated D‑dimer level, and a reduced fibrinogen level that allowed to establish the diagnosis of kaposiform lymphangiomatosis (KLA). Sirolimus was implemented, and slight improvement was observed over the following years.
The spectrum of pulmonary lymphatic disorders includes macro- and microlymphatic malformations, primary and secondary lymphangiectasias, generalized lymphatic anomalies (previously known as diffuse pulmonary lymphangiomatoses), and lymphangioleiomyomatosis.1 Micro- or macrocystic lymphatic malformations are evident in patients with generalized lymphatic anomalies.1,2 Histological and immunohistological investigations reveal irregular, thin‑walled vascular channels lined by endothelial cells; which resemble lymphatic vessels in that they express D2‑40, PROX1, and LYVE‑1 (lymphatic vessel endothelial hyaluronan receptor 1).1,2
Recently, KLA has been described on the basis of specific clinical, radiological, and histological features.2 Although the disease principally presents in childhood (regardless of sex), a few adult cases have been diagnosed.1-3 The clinical presentation is variable, ranging from a localized disease to generalized lymphatic anomalies involving the lungs (80%), mediastinum (100%), bones (70%–50%), spleen (50%), soft tissue, or skin (35%).1-5 The characteristic abnormalities include hemoptysis, variable thrombocytopenia, and rapidly progressive, hemorrhagic lymphatic effusions in the pleural (85%), pericardial, or peritoneal cavities.1-5 Laboratory abnormalities usually reflect coagulopathy. The etiology and pathogenesis of KLA are poorly understood. The disease presents features of both lymphatic anomalies and neoplasia.2
The optimal treatment of KLA remains unknown Symptomatic treatment of the pleural effusion (thoracentesis, pleurodesis, thoracic duct ligation, pleuroperitoneal shunting, corticosteroids, vincristine, interferon α, or radiotherapy) is not effective.1,2,5 Recently, patients with KLA have been treated with mammalian target of rapamycin inhibitors, other antiangiogenic agents (such as bevacizumab), or PD‑L1 inhibitors.2,5
Patients exhibiting progressive pulmonary involvement often have poor prognosis (the 5‑year survival rate is observed in about 50%). Usually, childhood onset is associated with a more aggressive disease course.
- Luisi F, Torre O, Harari S. Thoracic involvement in generalized lymphatic anomaly (or lymphangiomatosis). Eur Respir Rev. 2016; 25: 101‑103. | Crossref
- Croteau SE, Kozakewich HP, Perez‑Atayde AR, et al. Kaposiform lymphangiomatosis: a distinct aggressive lymphatic anomaly. J Pediatr. 2014; 164: 383‑388. | Crossref
- Shah V, Shah S, Barnacle A, et al. Mediastinal involvement in lymphangiomatosis: a previously unreported MRI sign. Pediatr Radiol. 2011; 41: 985‑992. | Crossref
- Yoo SH, Song JS, Lee JJ, et al. Diffuse pulmonary lymphangiomatosis: pulmonary lymphatic disorder in an adult. Basic and Applied Pathology. 2012; 5: 63‑66. | Crossref
- Safety and Efficacy Study of Sirolimus in Complicated Vascular Anomalies. ClinicalTrials.gov website. http://clinicaltrials.gov/show/NCT00975819. Accessed January, 2015.
ARTICLE INFORMATION