Coincident relapsing polychondritis and IgG4–related disease: a diagnostic challenge



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Coincident relapsing polychondritis and IgG4–related disease: a diagnostic challenge
Introduction
Relapsing polychondritis (RP) and immunoglobulin G4–related disease (IgG4‑RD) are rare multi‑organ immune‑mediated diseases. Both can present with nonspecific symptoms of fatigue, weight loss, arthralgia, raised inflammatory markers, or chronic anemia, and diagnostic delay is common. Early recognition and diagnosis are important to minimize morbidity and risk of incapacity as both diseases have a potentially good treatment response.1,2
Relapsing polychondritis is characterized by recurrent episodes of inflammation of cartilage‑rich tissues, typically affecting the ear, nose, or tracheobronchial tree. First presenting as recurrent and self‑limiting episodes of acute inflammation, the disease causes tissue destruction and functional impairment, such as deafness and respiratory tract stenosis if left untreated.1,3 IgG4‑RD is associated with clinical or radiological masses that can appear in almost every organ, but predominantly in the salivary and lachrymal glands, lymph nodes, pancreas, lungs, and kidneys. The diagnosis is suspected from clinical and radiological examination findings and confirmed by histology of the involved tissues, typically showing a combination of lymphoplasmacytic infiltrates with a high count of IgG4‑positive plasma cells, storiform fibrosis, and obliterative phlebitis. However, histological diagnostic criteria vary depending on the organ, and mostly 2 of the 3 cornerstone findings are present.2,4
Case report
A 78‑year‑old man with a past medical history of type 2 diabetes, hypertension, dyslipidemia, and an episode of pancreatitis 1 year earlier presented in June 2016 with a 6‑month history of extensor tenosynovitis and arthritis of the left wrist, accompanied by fatigue, weight loss, and Raynaud phenomenon. He had iron deficiency anemia, (hemoglobin, 96 g/l; reference range, 130–180 g/l), raised C‑reactive protein level (175 mg/l; reference range <5 mg/l), and antinuclear antibodies titer of 1/160, as well as emphysematous pulmonary changes and myelodysplasia with no excess of blasts on bone marrow biopsy.
In 2017, he was admitted to the hospital twice because of perichondritis of left ear accompanied by vertigo, hoarse voice, nasal tenderness, and erythema nodosum. Positron emission tomography (PET) and computed tomography (CT) showed increased fluorodeoxyglucose (FDG) uptake in the right posterior nasopharynx wall and 2 hypermetabolic lung consolidations in the right upper and lower lobe, but no uptake in the cartilage of the ears, nose, or tracheobronchial tree (Figure 1A). Prednisolone 30 mg/d was initiated for an undetermined autoimmune disorder. The weight, fatigue, arthritis, anemia, and inflammatory markers improved quickly and prednisolone was tapered to 15 mg/d. Following subsequent admissions with cough, dyspnea, and raised inflammatory markers, chest CT scan was performed and showed bilateral lung consolidations with features of organizing pneumonia, traction bronchiectasis, mediastinal adenopathy, and bilateral peripheral pulmonary embolism (Figure 1B). Pseudomonas aeruginosa and Klebsiella pneumoniae were identified in bronchoalveolar lavage fluid.
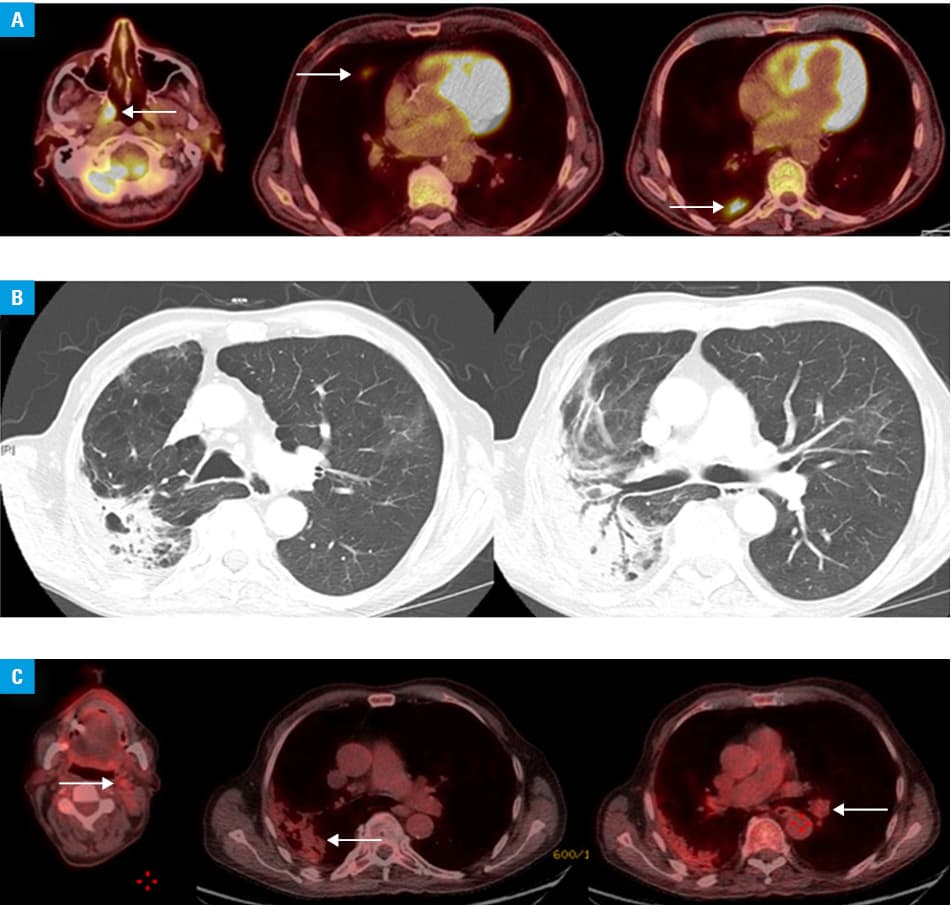
In 2018, after reducing the dose of prednisolone to 10 mg/d the patient developed deafness and vestibular neuronitis with no cause identified at the time. Further presentation with acute kidney injury (serum creatinine, 246 µmol/l) led to a kidney biopsy revealing tubular interstitial inflammation with early fibrosis, IgG4 positive lymphoplasmocytes (10% to 15% per high‑power field), and IgG4 positive staining of the tubular basement membrane. Serum IgG4 levels were raised at 481 mg/dl (normal values <135 mg/dl). PET‑CT showed positive FDG uptake in the right lower lobe of the lung, in a left cervical lymph node, and hila consistent with IgG4‑related disease (Figure 1C).
On referral to a tertiary center, he was noted to have erythema nodosum, left submandibular sialadenitis, C‑reactive protein level of 100 mg/l, and hemoglobin level of 100 g/l. Prednisolone was increased to 15 mg/d and rituximab 1000 mgat day 0 and day 14 commenced. Following this, significant improvement was noted and prednisolone could be tapered below 7 mg/d. His inflammatory and constitutional symptoms resolved, his renal function came back to baseline with a serum creatinine of 130 µmol/l, and his IgG4 level fell below 250 mg/dl.
Discussion
We have presented a 78‑year‑old man with features of both RP and IgG4–related disease (IgG4‑RD). There was a lengthy period of diagnostic uncertainty that delayed effective therapy.
He fulfilled McAdam, Damiani, Levine, and Michet diagnostic criteria for RP by presenting with at least 3 clinical features of RP: auricular chondritis, nonerosive seronegative polyarthritis and sensorineural hearing loss, and a positive response to corticosteroids administration. In addition, he had constitutional symptoms and erythema nodosum.1,3 At the same time, there were features of IgG4‑RD present: extensor enthesopathy, pancreatitis, lung consolidations, submandibular sialadenitis, posterior nasopharynx PET‑FDG uptake, hilar and cervical lymphadenopathy, high IgG4 serum titers, tubulointerstitial nephritis with lymphoplasmacytic infiltrates, fibrosis, and high counts of IgG4‑positive plasma cells. He also fulfilled the latest diagnostic criteria for definite IgG4‑RD because of the presence of localized masses or swellings in multiple organs (lymph nodes, lungs, submandibular gland, kidney), serum IgG4 concentrations above 135mg/dl (481 mg/dl), and a histologic confirmation with lymphoplasmacytic infiltrates, fibrosis, and more than 10 IgG4‑positive plasma cells per high‑power field.2,4
It is difficult to establish with confidence that the episodes of pulmonary exacerbations were due to IgG4‑RD and not RP as both diseases can present with shortness of breath, cough, and increased sputum production. Lung involvement in IgG4‑RD consists of nonspecific interstitial pneumonia, organizing pneumonia (as in the presented case), or pulmonary consolidations (as in the presented case).2,4 In RP, patients usually present with acute respiratory distress caused by tracheobronchial chondritis or with symptoms of pneumonia due to infections triggered by secondary tracheobronchomalacia and bronchiectasis.1,3 For our patient, no CT scan–reported signs of chondritis and no pathogenic germs were identified by sputum or bronchoalveolar lavage; Pseudomonas aeruginosa and Klebsiella pneumoniae being commensal bacteria found in the flora of healthy individuals.
Moreover, the patient’s kidney involvement, interstitial nephritis, was typical of IgG4‑RD, and not RP. In RP kidney involvement is rare and usually presents as mesangial expansion, IgA nephropathy, or crescentic glomerulonephritis.1-4
There are 2 explanations for this patient’s poor response to steroid treatment. First, current recommendations suggest an initial prednisolone dose of 0.6 to 1 mg/kg/d for IgG4‑RD and 0.25 to 1 mg/kg/d for RP for 2 to 4 weeks before gradual tapering and a maintenance dose higher than 20 mg/d usually for 2 to 6 months.1-4 Our patient weighted 80 kilograms and received an initial dose of 0.375 mg/kg/d (30 mg/d) for 4 weeks and a dose above 0.25 mg/kg/d (20 mg/d) for 6 weeks in total – not reaching currently recommended targets. Second, he had all the risk factors for refractory RP disease and poor prognosis: male sex, age at diagnosis above 55 years, myelodysplasia, cutaneous involvement, and general symptoms.5
Even though RP and IgG4‑RD respond well to corticosteroid therapy, treatment for corticoresistant or corticodependent cases differ.1,2 For relapsing polychondritis, azathioprine, methotrexate, cyclophosphamide, or infliximab are mostly used and rituximab is controversial.1,3 In IgG4‑RD, corticoresistant cases are usually treated with azathioprine, mycophenolate mofetil, methotrexate, or rituximab.2,4 We made the choice to treat our patient with rituximab and his good response reinforces the rationale for rituximab in patients with RP and IgG4‑RD.
The range of features and disease associations of IgG4‑RD continue to expand, while RP is associated with another autoimmune disease in 30% of cases. However, co‑ocurence of RP and IgG4 has only been reported in 2 cases where the diagnosis of RP preceded the diagnosis of IgG4‑RD by 20 years and 14 months and symptoms resolved with steroid treatment.1,6 Our case is the first description of concomitant occurrence of RP and IgG4‑RD and the first patient who responded well to treatment with rituximab.
Finally, in this case, one could question the presence of RP and suggest instead that only IgG4‑RD was present with some atypical RP‑like symptoms. IgG4‑RD is described as a “great mimicker” and can present with nasal, sinusal, orbital, periorbital, salivary, or lachrymal inflammatory masses.7 However, to the best of our knowledge, no case of IgG4‑RD presenting with perichondritis of the ears has been described so far, except for the 3 cases mentioned here above in which this presentation was attributed to RP and not IgG4‑RD. One of these patients having undergone an ear biopsy that discarded IgG4 plasmocyte infiltration, fibrosis, or venous embolism in the ears.6 Moreover, our patient fulfilled not one but all 3 existing diagnostic criteria for RP. He presented with skin disease typical for RP and not IgG4‑RD and with myelodysplasia that is typically associated with RP. Bone marrow biopsy revealed no fibrosis, venous thrombosis, or IgG4 lymphoplasmacytic infiltrate.
To conclude, in complex multisystem presentations of inflammatory disease, a broad diagnostic approach is required. With further understanding of pathogenic mechanisms and identification of newer phenotypes, traditional approaches to definition and classification of multisystem disease are being questioned. This represents a challenge for the medical community to collate large scale cohorts and continue to refine classification and diagnostic pathways.
Lessons to be learned
The following learning points can be emphasized from this case report: a) Our patient presented with clear features of RP and IgG4‑RD once different presentations over preceding 3 years were put together; b) repeated admissions for bronchitis or pneumonia in patients without prior history of chronic bronchitis and without isolation of pathogenic germs should raise suspicion for a possible underlying autoimmune disease; c) the need to refer patients with rare autoimmune conditions or repeated unexplained presentations to reference centers; d) the necessity for steroids at adequate doses for treatment of RP and IgG4‑RD; e) the benefit of rituximab in RP, IgG4‑RD, or in cases where both diseases are present together
- Borgia F, Giuffrida R, Guarneri F, Cannavò SP. Relapsing polychondritis: an updated review. Biomedicines. 2018; 6: 84. | Crossref
- Kamisawa T, Zen Y, Pilai S, Stone H. IgG4‑related disease. Lancet. 2015; 385: 1460‑1471. | Crossref
- Mathian A, Miyara M, Cohen‑Aubart F, et al. Relapsing polychondritis: a 2016 update on clinical features, diagnostic tools, treatment and biological drug use. Best Pract Res Clin Rheumatol. 2016; 30: 316‑333. | Crossref
- Hamano H, Tanaka E, Ishizaka N, Kawa S. IgG4‑related disease ‑ a systemic disease that deserves attention regardless of one’s subspecialty. Intern Med. 2017; 57: 1201‑1207. | Crossref
- Dion J, Costedoat‐Chalumeau N, Sène D, et al. Relapsing polychondritis can be characterized by three different clinical phenotypes: analysis of a recent series of 142 patients. Arthritis Rheumatol. 2016; 68: 2992‑3001. | Crossref
ARTICLE INFORMATION