In March 2019, a 35‑year‑old man with obesity was referred to the hospital due to progressive weakness, laboratory abnormalities (anemia, leukopenia, increased serum creatinine levels), and fever (reduced by steroid treatment). His complaints started in 2006 with pneumonia, high fever accompanied by chills, and arthralgia of unknown origin. He had no family history of kidney or connective tissue diseases. Laboratory workup revealed seropositivity for antinuclear and anti–double‑stranded DNA antibodies, leukopenia, thrombocytopenia, and hypergammaglobulinemia. Another episode of fever, arthralgia, anemia, leukopenia, and massive proteinuria (9 g/d) occurred in 2013. Between 2013 and 2019, due to recurrent fever, the patient periodically took glucocorticoids (prednisolone, 5 mg/d) prescribed by a general practitioner without consultation with a rheumatologist.
On examination, the patient was pale and had swollen lower extremities. His blood pressure was 160/90 mm Hg and heart rate was 110 bpm. Sonography revealed hepatosplenomegaly and portal hypertension. The length of the right and left kidney was 130 mm and 126 mm, respectively, and both organs showed marked hyperechogenicity. Laboratory examination indicated impaired renal function (serum creatinine, 2.53 mg/dl; estimated glomerular filtration rate, 30 ml/min/1.73 m2), proteinuria of up to 3.8 g/d, anemia (hemoglobin, 7 g/dl), a white blood cell count of 4710/µl, and a platelet count of 14.8 × 104/µl. Serum complement components were normal (C3, 141 mg/dl [reference range, 90–180 mg/dl; C4, 35 mg/dl [reference range, 10–40 mg/dl]). The patient tested positive for antinuclear and anti‑ribosomal P protein antibodies (titer, 1:3200), but negative for anti–double‑stranded DNA and anticardiolipin antibodies. He had decreased levels of serum albumin (2.5 g/dl) and slightly elevated levels of C‑reactive protein (39 mg/l).
The patient’s clinical presentation was typical of lupus nephritis (he met the American Rheumatism Association criteria for systemic lupus erythematosus [SLE] both in 2006 [≥6] and 2013 [≥5]). During hospitalization at our department, renal biopsy was performed, followed by an immediate intravenous administration of methylprednisolone (500 mg/d for 3 days), and then by oral prednisone. Surprisingly, histologic examination showed no features of lupus nephritis but was suggestive of amyloidosis (Figure 1A–1D). Finally, the patient discontinued glucocorticoid therapy and underwent additional tests, which allowed for the diagnosis of AA amyloidosis. Plasma cell dyscrasia was excluded based on the results of peripheral blood flow cytometry (CD19+ B cells, CD20+ B cells, lambda/kappa ratio of 1.21).
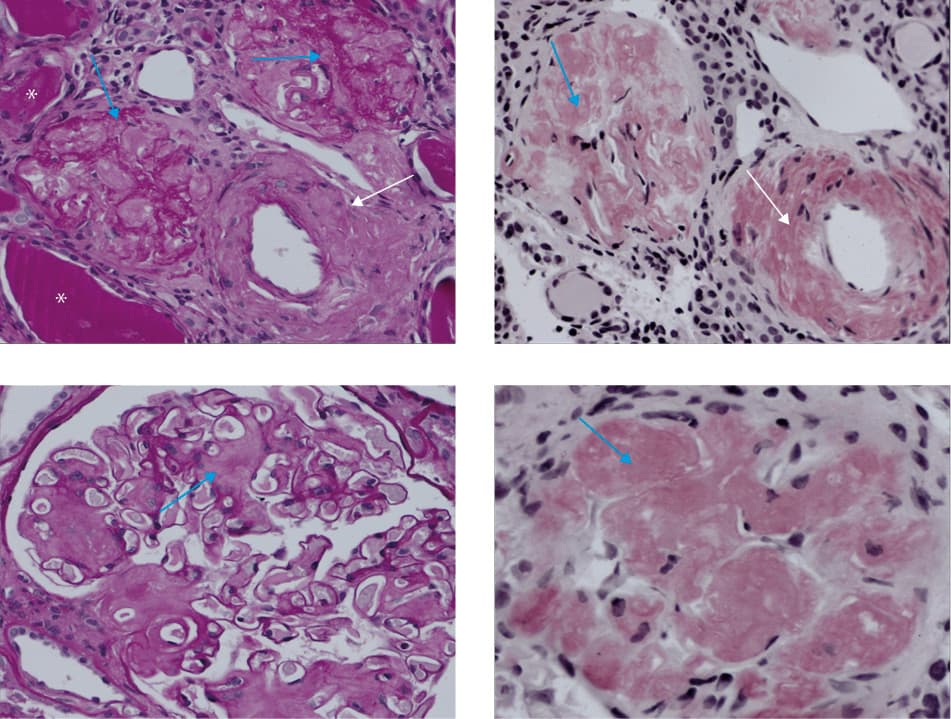
Amyloidosis is a well‑known complication of chronic inflammatory and connective tissue diseases. However, its association with SLE is extremely rare (there have been fewer than 40 cases reported since 1956).1,2 One of the causes may be a low level of serum amyloid A in the course of active lupus (not measured in our patient).3 It seems that a persistent, long‑lasting elevation of the serum amyloid A is needed for the development of AA amyloidosis.4
In this case, AA amyloidosis developed acutely with the symptoms of nephrotic syndrome in a patient who was finally diagnosed with SLE, but with a 13‑year delay. In our opinion, the major reason for the development of amyloidosis was long‑lasting inflammation, treated ineffectively with glucocorticoids in an undiagnosed patient. Of note, the period of time between the diagnoses of SLE and amyloidosis may vary from 1 year to 35 years.2 We suggest that amyloidosis, although uncommon, should be considered in the differential diagnosis of proteinuria in patients with SLE.
- Monteiro P, Abreu P, Salvador M, Malcata A. Secondary amyloidosis and systemic lupus erythematosus . Acta Rheumatol Port. 2009; 34: 400‑404.
- Rezgui A, Hassine IB, Karmani M, et al. Amyloïdosis, sarcoidosis and systemic lupus erythematosus. Pan African Medical Journal. 2016; 24: 23. | Crossref
- Ridley MG, Maddison PJ, Tribe CR. Amyloidosis and systemic lupus erythematosus. Ann Rheum Dis. 1984; 43: 649‑650. | Crossref
- Benson M, Cohen A. Serum amyloid A protein in amyloidosis, rheumatoid arthritis and neoplasmatic disease. Arthritis Rheum. 1979; 22: 36‑42. | Crossref
ARTICLE INFORMATION