To the editor
Left ventricular hypertrophy (LVH) is common in patients with cardiovascular disease, in those with cardiovascular risk factors, and in healthy individuals. Although electrocardiography is a first‑line diagnostic modality, echocardiography is highly recommended to confirm the diagnosis. According to recent guidelines, LVH is defined as an increase of left ventricular (LV) mass, with the cutoff value of more than 150 g (>90 g/m2) for women and more than 200 g (>103 g/m2) for men. In addition, septal and posterior wall thickness should exceed 9 mm and 10 mm for women and men, respectively.1 At the microscopic level, LVH is characterized by an increase in the size or number of sarcomeres (or both).
Although common, LVH should not be regarded as an insignificant marker of disease. On the contrary, a thorough diagnostic workup should be performed once it is detected. A mere simplification by connecting LVH with hypertension may result in the underdiagnosis of a substantial number of patients and, consequently, may lead to ineffective and even detrimental therapy. Since it is highly impractical to simply list the multiple conditions that may be associated with LVH, what is needed is a comprehensive LVH diagnostic algorithm as a way to facilitate a differential diagnosis. In line with this, a simplified 3‑stage workup has been developed (Figure 1). In brief, the physiologic causes of LVH should be considered first, followed by the most common conditions that cause LVH. Hypertrophic cardiomyopathy (HCM) is a natural consequence of LVH; however, other rare causes of LVH are also possible. A detailed description of the numerous conditions leading to LVH is beyond the scope of this paper; nevertheless, certain useful pointers and red flags were briefly highlighted below.
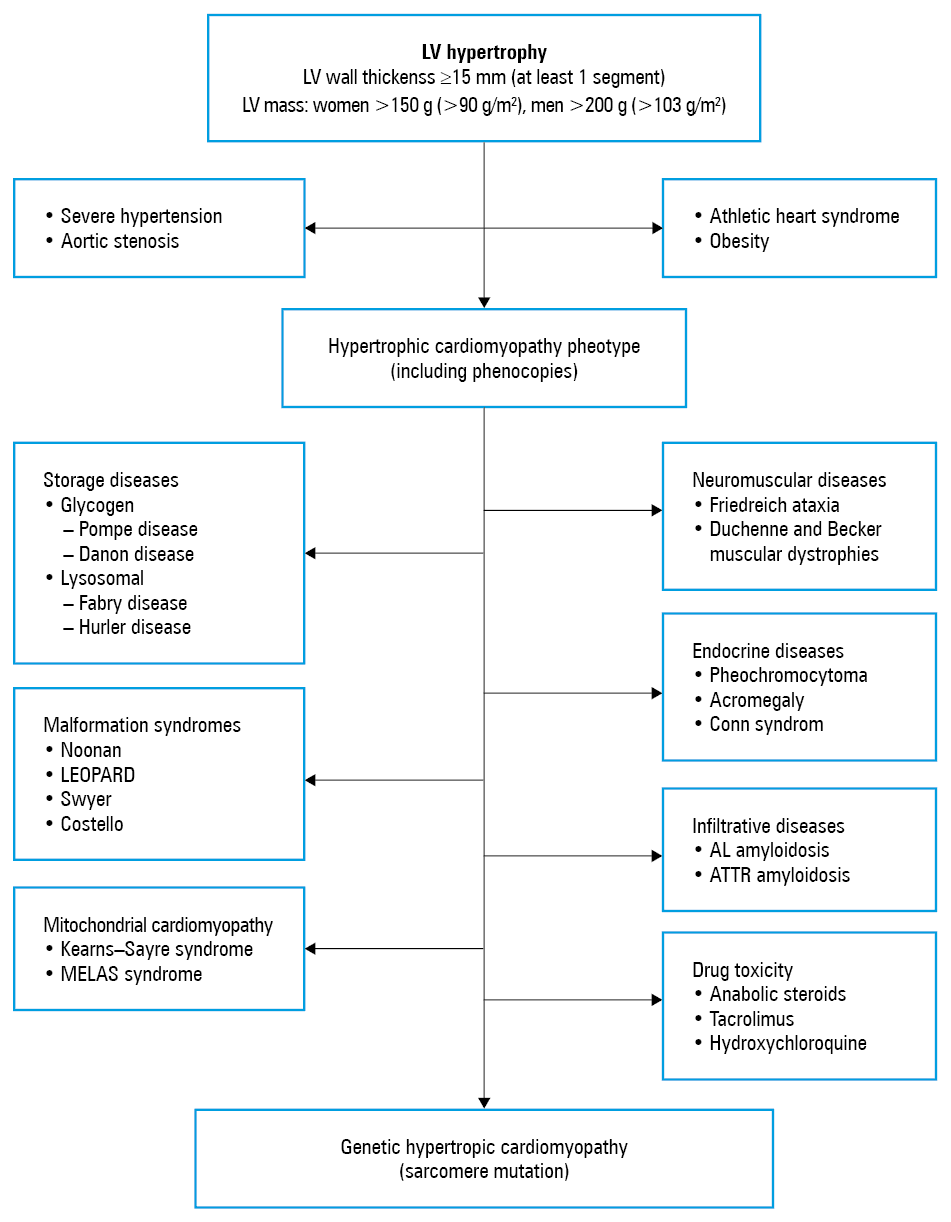
Generally, the cause of LVH nay be either physiologic or pathological. The physiologic causes occur in 3 distinct groups of people: athletes, overweight or obese individuals, and pregnant women.2 Typically, highly‑trained athletes may develop LVH, known as the “athlete’s heart,” but apart from a history of long‑term training, the following red flags should be considered: a moderate LVH of 13 to 15 mm, increased LV cavity size, concentric LVH, no family history of LVH, or at least 2‑mm regression of the wall thickness after detraining.
As for pathological causes, the most common pathomechanism of LVH is a long‑term increase in afterload, as observed in hypertension or aortic stenosis. Untreated or poorly controlled hypertension results in mild‑to‑moderate LVH (usually <15 mm) and is frequently accompanied by other features, such as history of hypertension or evidence of other end‑organ damage.3 Moderate concentric LVH is present in the majority of patients with degenerative aortic stenosis.4 Less commonly, LVH results from volume overload states, such as in aortic and mitral regurgitation, dilated or restrictive cardiomyopathy, heart failure, ventricular septal defect, and aortic coarctation.5-7
Another relatively common condition that is always associated with LVH is HCM. In fact, HCM is not a rare condition as its prevalence can vary between 0.2% (1 of 500 people/population) and 0.5% (1 of 200 people/population), which makes it the most widespread cardiomyopathy.8 To be definitive on the nature of HCM and the terminology used here, it should be clearly stated that HCM is a genetic disease caused by mutations in cardiac sarcomere protein genes, whereas all other conditions that phenotypically mimic HCM are not HCM as such but are instead termed phenocopies.9
Hypertrophic cardiomyopathy is inherited in an autosomal dominant pattern with variable expressivity and age‑related penetrance. So far, more than 1500 (often private) mutations in 11 genes that encode thick or thin myofilament proteins of the sarcomere have been associated with HCM. These mutations result in hypertrophied and disorganized cardiomyocytes. Hypertrophic cardiomyopathy is characterized by LVH of various degrees, morphologies, and patterns: LV wall thickness exceeding 15 mm in any myocardial segment and asymmetric septal hypertrophy (the ratio of septal to posterior wall thickness >1.3 for normotensive individuals or >1.5 for hypertensive patients) are all considered diagnostic for HCM. The interventricular septum is typically affected, and its morphology may vary from reverse curvature, neutral, to sigmoid. Asymmetric LVH of the basal anterior septum is most common, followed by that of the anterior free wall and posterior septum; however, any localization, including concentric, mid‑ventricular and apical, is possible.
While most patients with HCM have no or just minimal symptoms, a substantial proportion of individuals (15%–20%) complain of a variety of symptoms, such as chest pain, palpitations, fatigue, dyspnea, and presyncopal states or syncope. These clinical manifestations result from various pathologies that can often coexist, such as diastolic dysfunction, microvascular angina, LV outflow tract obstruction, supraventricular arrhythmias (most frequently atrial fibrillation) and ventricular arrhythmias (that may lead to sudden cardiac death), stroke as a result of atrial fibrillation but also caused by intrinsic blood coagulation disorders, and the end‑stage phase of HCM, which leads to heart failure.10-12
Finally, in approximately 5% to 10% of patients with LVH, who would otherwise be diagnosed with HCM, much rarer causes warrant further investigation. Unlike in HCM or hypertension, LVH in those rare conditions does not always result from cardiomyocyte hypertrophy but rather from intra- or extracellular deposits. Among the storage diseases, glycogen and lysosomal diseases are most prominent. Glycogen diseases arise from inborn errors of glycogen metabolism leading to the accumulation of various glycogen byproducts. Deficiency in acid maltase (Pompe disease) and in lysosome‑associated membrane protein 2 (Danon disease) is known to cause severe LVH. Although the diagnosis of a glycogen storage disease is definitely not straightforward, there are some signs that can be considered red flags, such as progressive myopathy accompanied by a high level of creatine kinase, hypotension, ophthalmologic abnormalities, and variable intellectual disability. Currently, enzyme replacement therapy (ERT) is available only for Pompe disease. Among the lysosomal diseases, Fabry disease is an X‑linked storage disease that is caused by deficient activity of the lysosomal enzyme alpha‑galactosidase A, resulting in the accumulation of globotriaosylceramide in lysosomes in multiple cell types in the body. Patients present with peripheral neuropathy of the hands and feet, alimentary tract disorders (nausea, abdominal pain, diarrhea), progressive renal insufficiency, reduced sweating and the cutaneous lesions of angiokeratoma corporis, corneal deposits, and cerebrovascular involvement (transient ischemic attack and stroke). As for cardiovascular manifestations, LVH, conduction disturbances (usually pacemakers are indicated), and hypertension are most often seen. The detection of low or no alpha‑galactosidase activity is confirmatory for the diagnosis of Fabry disease. Establishing the correct diagnosis is important since ERT therapy (agalsidase alfa and beta) has been shown to reduce LVH and ameliorate renal failure.13
Another group of diseases that are frequently associated with LVH are infiltrative diseases, including amyloidosis, sarcoidosis, and hemochromatosis. Amyloidosis is a heterogeneous group of diseases characterized by the deposition of amyloid fibrils. Until now, more than 30 different misfolded and/or misassembled proteins have been shown to have the propensity to form these types of amyloid fibrils. However, the ones that are most cardiac relevant are immunoglobulin light chain (AL amyloidosis) and transthyretin (ATTR amyloidosis). The clinical course and survival in cardiac amyloidosis are extremely poor. The survival is less than 2 years for AL amyloidosis and 2 to 3 years for ATTR amyloidosis. The diagnosis requires a multidisciplinary approach as well as the integration of various modalities such as 99mTc‑DPD scintigraphy, magnetic resonance imaging, bone marrow or endomyocardial biopsy, and laboratory testing, including genetic studies. The diagnosis should not be delayed as specific therapies are available for AL amyloidosis: multidrug chemotherapy and bone marrow transplantation, whereas for ATTR amyloidosis, there are a number of drugs available in clinical practice, including tafamidis, doxycycline, or patisiran.14,15
Rare multisystemic malformation syndromes, such as Noonan, Costello, LEOPARD, or Swyer, are also associated with LVH. In most instances, the correct diagnosis is established in childhood. Dysmorphic features, such as short stature, hypertelorism, lower ear positioning, hypermobility of the joints, chest deformation, and various degrees of mental retardation should raise suspicion. In mitochondrial diseases, such as Kearns–Sayre or MELAS syndromes, LVH is also a common feature. Red flags such as myopathy (raised creatine kinase levels), deafness, visual disturbances, hypotension, and metabolic acidosis (raised lactate levels) may aid the diagnosis. Left ventricular hypertrophy can also be observed in neuromuscular disorders, such as Duchenne and Backer muscular dystrophies or Friedreich ataxia. These are severe neurologic conditions that usually lead to advanced physical disability and respiratory failure. However, due to progress in neurologic and pulmonary rehabilitation, including the availability of home respirators, some of those patients gradually develop heart failure due to dilated or hypertrophied hearts. In some rare endocrine disorders, such as pheochromocytoma, acromegaly, or Conn syndrome, LVH is a frequent symptom due to poorly controlled hypertension. Lastly, LVH may result from drug toxicity, particularly with the use of anabolic steroids or tacrolimus.
It is not clear whether the list of potential factors conducive to LVH is complete; nevertheless, a more focused approach, including the consideration of rare diseases, may help raise awareness of the numerous conditions that may manifest with LVH.
- Lang RM, Badano LP, Mor‑Avi V, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging. 2015; 16: 233‑270.
- Skrypnik D, Bogdański P, Zawiejska A, Wender‑Ożegowska E. Role of gestational weight gain, gestational diabetes, breastfeeding, and hypertension in mother‑to‑child obesity transmission. Pol Arch Intern Med. 2019; 129: 267‑275. | Crossref
- Hering D, Trzebski A, Narkiewicz K. Recent advances in the pathophysiology of arterial hypertension: potential implications for clinical practice. Pol Arch Intern Med. 2017; 127: 195‑204. | Crossref
- Roleder T, Hawranek M, Gąsior T, et al. Trends in diagnosis and treatment of aortic stenosis in the years 2006‑2016 according to the SILCARD registry. Pol Arch Intern Med. 2018; 128: 739‑745.
- Ostręga M, Gierlotka MJ, Słonka G, et al. Clinical characteristics, treatment, and prognosis of patients with ischemic and nonischemic acute severe heart failure. Analysis of data from the COMMIT‑AHF registry. Pol Arch Intern Med. 2017; 127: 328‑335.
ARTICLE INFORMATION