Bradykinin and oxidative stress in patients with hereditary angioedema due to C1 inhibitor deficiency
Key words: antioxidant, bradykinin, C1 inhibitor, hereditary angioedema, oxidative stress



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Bradykinin and oxidative stress in patients with hereditary angioedema due to C1 inhibitor deficiency
Introduction: Hereditary angioedema (HAE) is a rare autosomal dominant disease caused by genetic dysfunction of C1 inhibitor (C1‑INH) due to mutations in the SERPING1 gene. The disorder is mediated mainly by bradykinin. The clinical course of the disease is varied and not related to genetic changes.
Objectives: We aimed to evaluate redox homeostasis of peripheral blood mononuclear cells (PBMCs) in patients with HAE due to C1‑INH deficiency (C1‑INH‑HAE) by measuring the levels of reactive oxygen species (ROS) of PBMCs as well as plasma advanced glycation end products (AGEs) and advanced oxidation protein products (AOPPs). We also aimed to assess the effect of bradykinin on ROS levels.
Patients and methods: We enrolled 30 adults with C1‑INH‑HAE and 15 healthy individuals. The levels of ROS were measured by flow cytometry, while the plasma levels of AGEs and AOPPs were determined spectrophotometrically by enzyme‑linked immunosorbent assays.
Results: Basal and hydrogen peroxide (H2O2)–induced ROS levels were higher in patients with HAE when compared with controls (P = 0.002 and P = 0.001, respectively), indicating abnormalities in redox homeostasis. Plasma AOPP and AGE levels were similar in both groups. Bradykinin reduced basal and H2O2-induced ROS generation in PBMCs only in patients with HAE (P = 0.03).
Conclusions: The higher basal and H2O2-induced ROS levels in patients with C1‑INH‑HAE indicate redox imbalance. However, by reducing basal and H2O2-induced ROS levels, bradykinin shows antioxidant action in this disorder.
What's new?
Hereditary angioedema due to C1‑inhibitor deficiency (C1‑INH‑HAE) is a rare hereditary disease, in which the deficiency of C1 inhibitor disturbs the homeostasis of numerous systems, metabolism, and immunity. We examined the markers of redox imbalance (basal and hydrogen peroxide [H2O2]–induced levels of reactive oxygen species [ROS], plasma advanced glycation end products, and advanced oxidation protein products) in patients with C1‑INH‑HAE types I and II. We also assessed the effect of exogenous bradykinin on ROS levels. Our results are groundbreaking in that they revealed a significant increase in basal and H2O2-induced ROS levels as well as antioxidant action of exogenous bradykinin in patients with C1‑INH‑HAE.
Introduction
Hereditary angioedema (HAE) due to C1 inhibitor (C1‑INH) deficiency (C1‑INH‑HAE) is a rare autosomal dominant disease caused by C1‑INH deficiency (type I, 85% of patients with low antigenic and functional plasma levels of C1‑INH) or C1‑INH dysfunction (type II, 15% of patients with normal or elevated plasma levels of C1‑INH and low functional levels of C1‑INH).1 C1 inhibitor is essential for numerous physiological processes, including complement and coagulation systems as well as fibrinolysis and the kinin system.1-5 The disease is caused by heterogeneous mutations within the C1‑INH gene (SERPING1).6,7 Despite considerable progress in understanding of the pathological mechanism of this disorder, a number of phenomena have not been fully elucidated.
One of the ongoing open questions is the lack of a correlation between the phenotype and genotype of patients with HAE.1,6,7 Also, despite a constantly low level and activity of C1‑INH, some patients may experience symptom‑free periods or suffer from angioedema.8 Another interesting aspect is related to the disease course, which is different among the individuals from the same family despite similar genetic changes in the SERPING1 gene.9 As indicated by previous studies, neither the type of genetic changes nor complement parameters allow a prediction of the clinical course of the disease.2,7,9
It is generally accepted that bradykinin is the main mediator inducing an angioedema attack in patients with C1‑INH‑HAE.2-5,10-12 Bradykinin is a tissue hormone generated by the kinin system as an inflammatory product of the coagulation system.1-3,10-12 It acts via bradykinin B2 receptor (B2R) and is very quickly inactivated. However, its metabolites act via actively formed bradykinin B1 receptors (B1R). Bradykinin exerts several physiological effects. It increases the permeability of capillaries, leading to local edema, warming, and erythema. Its vasodilating activity results in the release of 3 potent mediators: tissue plasminogen activator, prostacyclin, and endothelium‑derived vascular relaxing factor. Bradykinin‑mediated angioedema seems to occur in individuals with hereditary or acquired C1‑INH deficiency owing to an easy activation of the complement, plasma contact, and kinin system, as a result of uncontrolled overproduction of kallikrein and, in consequence, of bradykinin. There may be various direct reasons for the increase of serum bradykinin levels inducing angioedema in a given patient, which are often difficult to establish.
Patients with C1‑INH‑HAE report diverse nonspecific triggers inducing the attacks of edema, such as trauma or injury, pressure, effort, stress, or infection.1-3,13 Bradykinin‑mediated angioedema may be also caused by various metabolic products and environmental agents.8 These diversity of triggers in patients with C1‑INH‑HAE suggests the presence of reactions based on a feedback phenomenon, allowing the human body to adapt to the changing internal or external environment independently from the stimulus, as well as to maintain homeostasis, in which appropriate redox balance may play an essential role.
The aim of the study was to evaluate oxidative stress in patients with C1‑INH deficiency by measuring basal and hydrogen peroxide (H2O2)–induced levels of reactive oxygen species (ROS) in peripheral blood mononuclear cells (PBMCs) as well as by measuring the plasma concentrations of advanced glycation end products (AGEs) and advanced oxidation protein products (AOPPs). Moreover, we evaluated the effect of bradykinin on basal and H2O2-induced levels of ROS in vitro.
Patients and methods
The study included 30 patients (21 women and 9 men) with C1‑INH‑HAE types I and II, diagnosed and remaining under the care of the HAE Center in Krakow, Poland (Table 1). The diagnosis of C1‑INH‑HAE was established on the basis of patient and family history, examination during angioedema attacks, as well as reduced serum C1‑INH antigen levels and C1‑INH activity below 50% of the reference values according to international criteria.6 The exclusion criteria were the presence of concurrent diseases and medication use. Blood samples were obtained during remission (no attacks for at least 2 weeks). Clinical and laboratory characteristics of the study group are presented in Table 1.
Parameter | Value | |
Data are presented as median (Q1; O3) unless otherwise indicated.
a Reference range, 0.21–0.39 g/l
b Reference range, 70%–130%
c Reference range, 0.1–0.4 g/l
Abbreviations: C1‑INH, C1 inhibitor; C4, complement component 4; fC1‑INH, functional activity of C1 inhibitor; HAE, hereditary angioedema | ||
Sex, n | Female | 21 |
Male | 9 | |
Family history, n | Yes | 15 |
No | 15 | |
HAE type, n | I | 24 |
II | 6 | |
Age, y | 37.5 (26; 53) | |
C1‑INHa, g/l | Type I | 0.06 (0.04; 0.069) |
Type II | 0.425 (0.22; 0.683) | |
fC1‑INHb, % | 22.3 (10.3; 26) | |
C4c, g/l | 0.049 (0.034; 0.067) | |
Symptom score, 0–3 | 2 (1.25; 3) | |
The severity of C1‑INH‑HAE was evaluated using a 3‑grade scale of symptom severity (0, asymptomatic; 1, mild [<6 attacks/year]; 2, moderate [6–12 attacks/year), and 3, severe [>12 attacks/year) (modified by Agostini et al).14
The biochemical diagnosis of C1‑INH‑HAE was established during the remission of symptoms and was based on the measurement of serum C1‑INH and complement component C4 levels as well as functional activity of plasma C1‑INH. Serum C1‑INH and C4 levels were measured by nephelometry with specific C1‑INH antiserum (Siemens Healthcare Diagnostics GmbH, Marburg, Germany), and the functional level of C1‑INH was determined using a functional chromogenic assay, Berichrom C1‑INH (Siemens Healthcare Diagnostics GmbH).
The control group included 15 healthy individuals (11 women and 4 men; mean age, 48 years; range, 30–61 years). All controls had a negative family history of HAE and normal serum levels of C1‑INH (median, 0.22 g/l [range, 0.21–0.29 g/l]; reference range, 0.21–0.39 g/l), functional activity of C1‑INH (median, 96.6% [range, 74%–119%]; reference range, 70%–130%), and serum C4 levels (median, 0.21 g/l [range, 0.18–0.3 g/l]; reference range, 0.1–0.4 g/l).
The study was approved by the Ethics Committee of Jagiellonian University in Krakow (104/B2014, 22 May 2014), and patients signed a written informed consent form. The study was carried out in accordance with the Declaration of Helsinki and its amendments.
Measurement of intracellular reactive oxygen species generation
Anticoagulant‑treated blood was collected in EDTA tubes and layered over a lymphocyte separation medium (Ficol, PAN‑Biotech GmbH, Aidenbach, Germany), enabling PBMC isolation by centrifugation in a density gradient medium (400 g, 30 minutes, 20°C). The isolated cells were rinsed with phosphate‑buffered saline (Gibco, Invitrogen, Paisley, United Kingdom) supplemented with 1% fetal bovine serum (Gibco) (400 g, 6 minutes, 4°C), and then counted using the Bürker chamber.
To detect ROS generation, flow cytometry with 2’,7’-dichlorodihydrofluorescein diacetate (DCFH‑DA) dye (Sigma‑Aldrich, St. Louis, Missouri, United States) was used, according to Wang et al15 and Sarkar et al.16 DCFH‑DA is a stable nonfluorescent and cell‑permeable compound, which is converted within the cell to nonfluorescent DCFH by intracellular esterases. The deesterified product is oxidized by intracellular ROS to the highly fluorescent 2’7’-dichlorofluorescein (DCF). The intensity of green fluorescence upon excitation at 488 nm is proportional to the intracellular level of ROS. For this assay, PBMCs isolated from blood were cultured in medium for 24 hours. Then, the cells were washed with Krebs–Ringer–Hepes (KRH) buffer, transferred into 2 tubes, and incubated with or without 100‑pM bradykinin in KRH buffer for 10 minutes. After that, both samples were loaded with 100-µM DCFH‑DA in KRH buffer. After incubation in 5% CO2/95% air at 37°C for 45 minutes in dark, the cells were washed, divided into 2 aliquots, and suspended in KRH buffer. The free radical generator H2O2 (1 μM) was added extracellularly to one sample for 10 minutes. Then, basal and H2O2-induced ROS levels were measured in PBMCs. DCF fluorescence was measured by flow cytometry (FACS Canto II, Becton Dickinson, San Jose, California, United States). Data were acquired and analyzed using the DIVA software (Becton Dickinson).
DCFH‑DA was dissolved in dimethyl sulfoxide (Sigma‑Aldrich) as the stock solution and kept frozen at a temperature of –20°C. For cell loading, DCFH‑DA from stock solution was mixed with KRH loading buffer (NaCl, 116 mM; KCl, 4 mM; MgCl2, 1 mM; CaCl2, 1.8 mM; glucose, 25 mM; HEPES acid, 10 mM; pH adjusted to 7.4) to the final concentration of 100 µM. Bradykinin (Bachem, Bubendorf, Switzerland) was dissolved in water as the stock solution and kept frozen at a temperature of –20°C until mixed with the loading buffer to the final concentration of 100 pM. H2O2 (Farmina, Kraków, Poland) was diluted in the loading buffer to the final concentration of 1 mM.
The plasma concentrations of AGEs and AOPPs were measured spectrophotometrically using commercially available enzyme‑linked immunosorbent assay kits (Bioassay Technology Laboratory, Shanghai, China) and a microplate reader (Multiskan, Termo Fisher Scientific, Waltham, Massachusetts, United States). Each sample was analyzed in duplicate.
Statistical analysis
The Shapiro–Wilk test was used to assess the normality of variable distribution, and the Levene test was applied to assess the homogeneity of variance. Data were presented as median and lower and upper quartiles (Q1; Q3). Box and whisker plots were generated with Statistics v13.0 (Statsoft Inc., Tulsa, Oklahoma, United States) and showed the median, interquartile range (IQR), and minimum and maximum values. The Mann–Whitney test was used to compare patient and control groups as well as to compare bradykinin‑treated and -untreated PBMCs. The significance level was set at a P value of less than 0.05. The Spearman rank correlation test was used to identify correlations between AOPP and AGE levels both in the study and control groups. Analyses were conducted using the Statistica software package version 13.0 (Statsoft Inc.).
Results
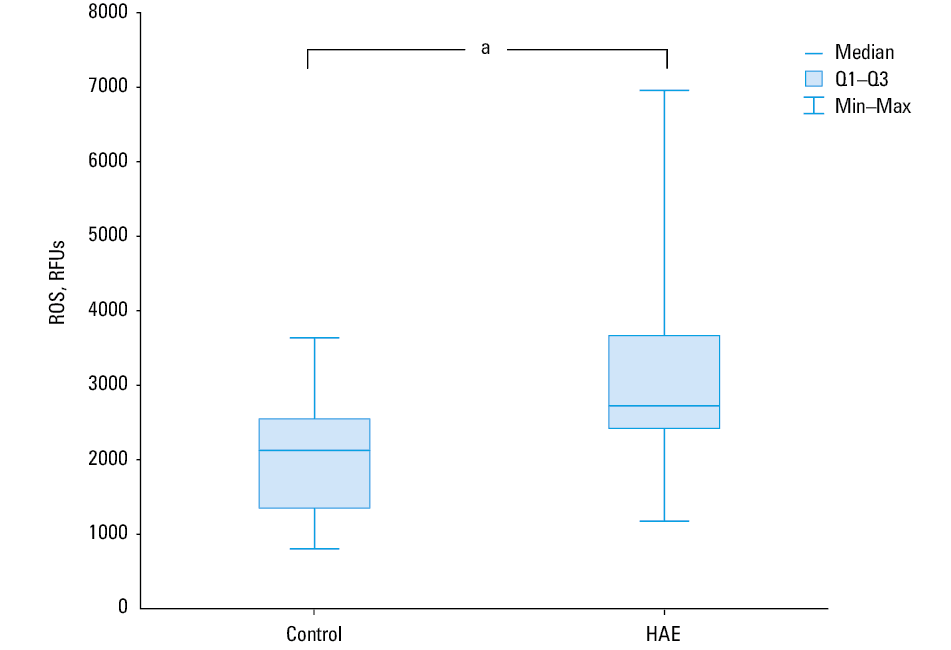
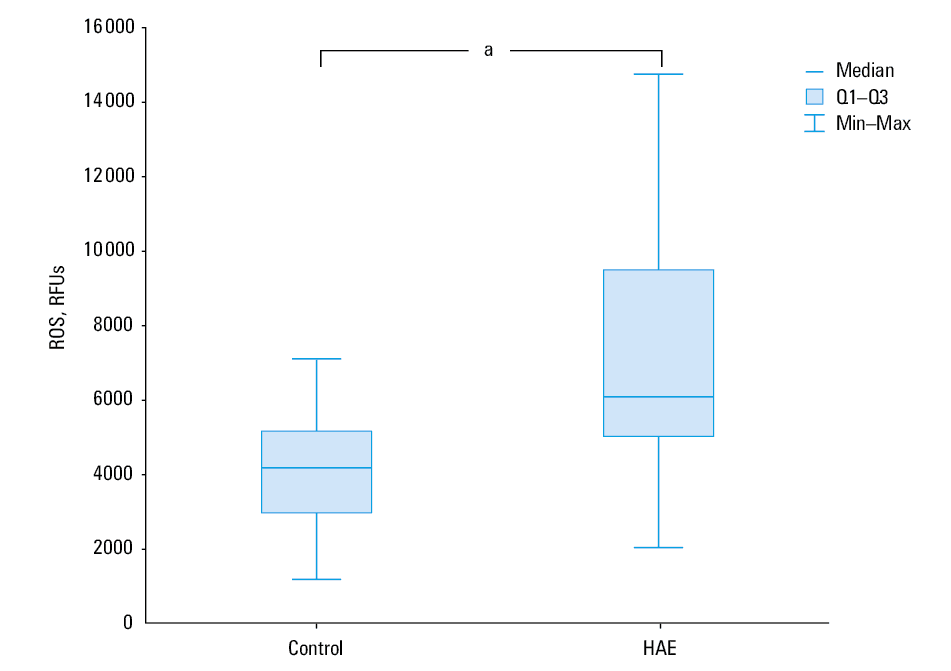
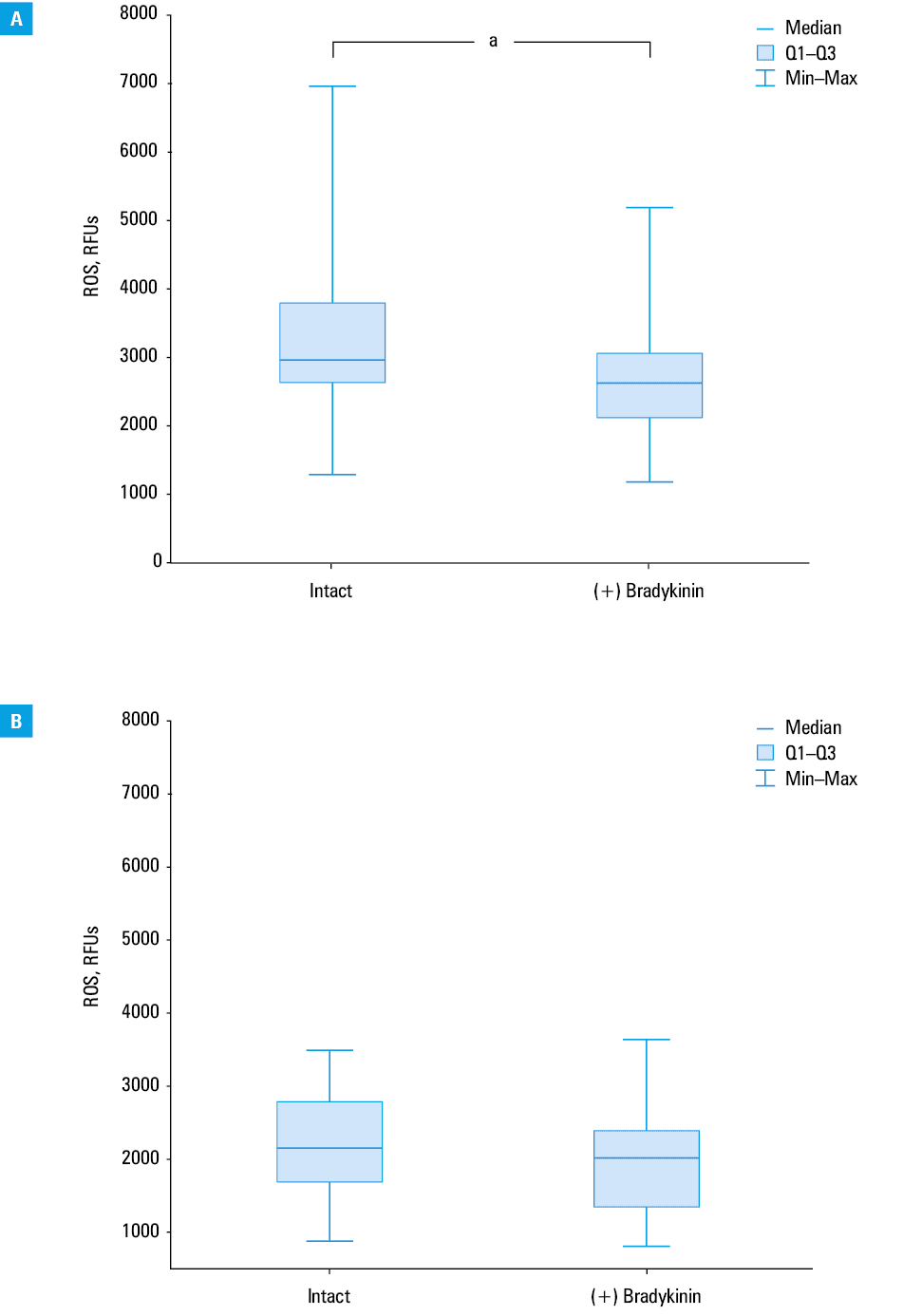
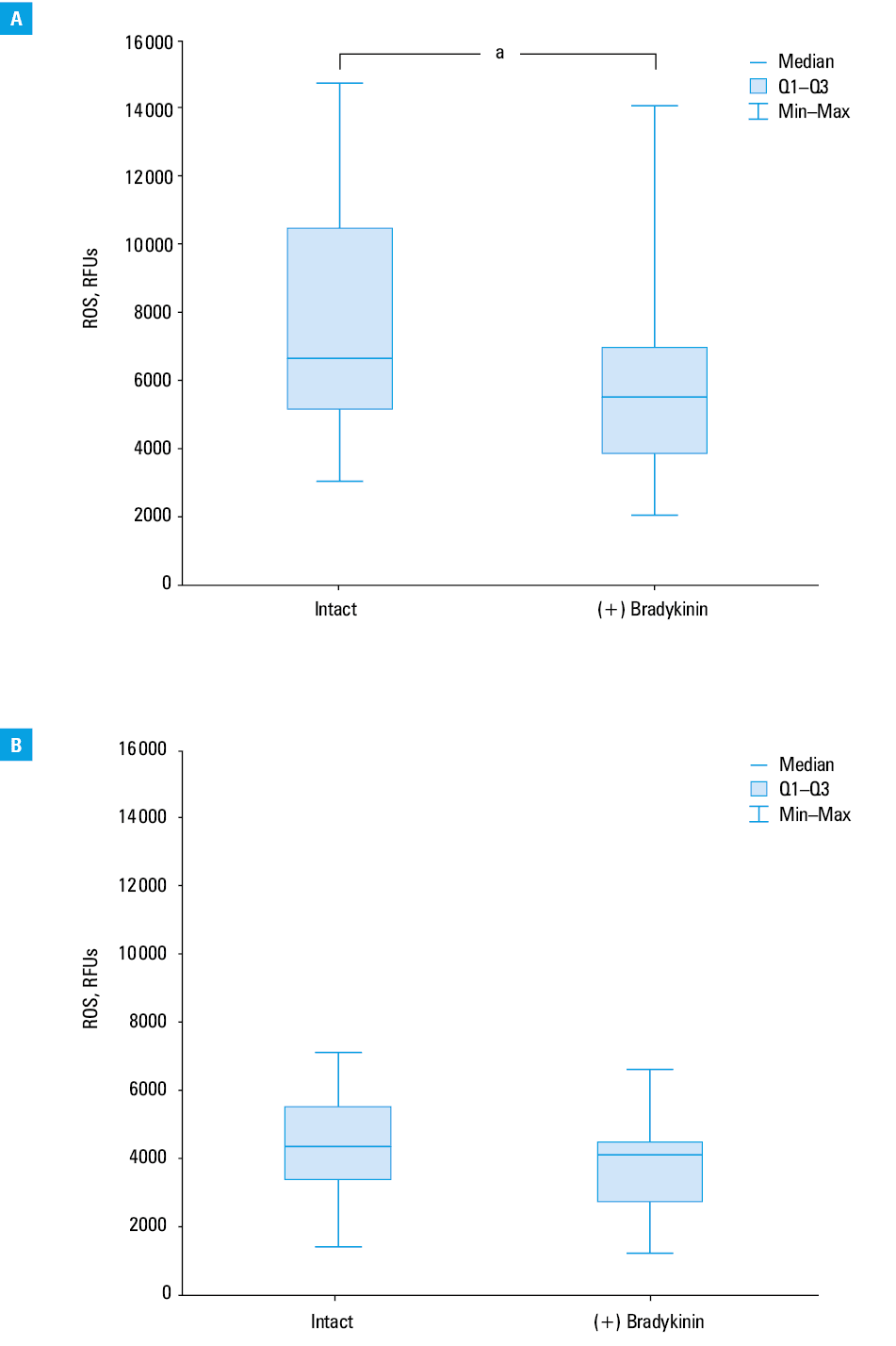
Basal ROS levels, expressed as DCF fluorescence intensity in relative fluorescence units (RFUs), were higher in PBMCs isolated from patients with HAE than in those obtained from controls (median [Q1; Q3], 2967 RFUs [2634; 3800] vs 2147 RFUs [1690; 2786]; P = 0.002; Figure 1). H2O2-induced ROS levels were also higher in PBMCs obtained from HAE patients than in those from healthy controls (median [Q1; Q3], 6672 RFUs [5 140; 10 494] vs 4352 RFUs [3390; 5531]; P = 0.001), indicating that patients with HAE have disturbances in redox homeostasis (Figure 2).
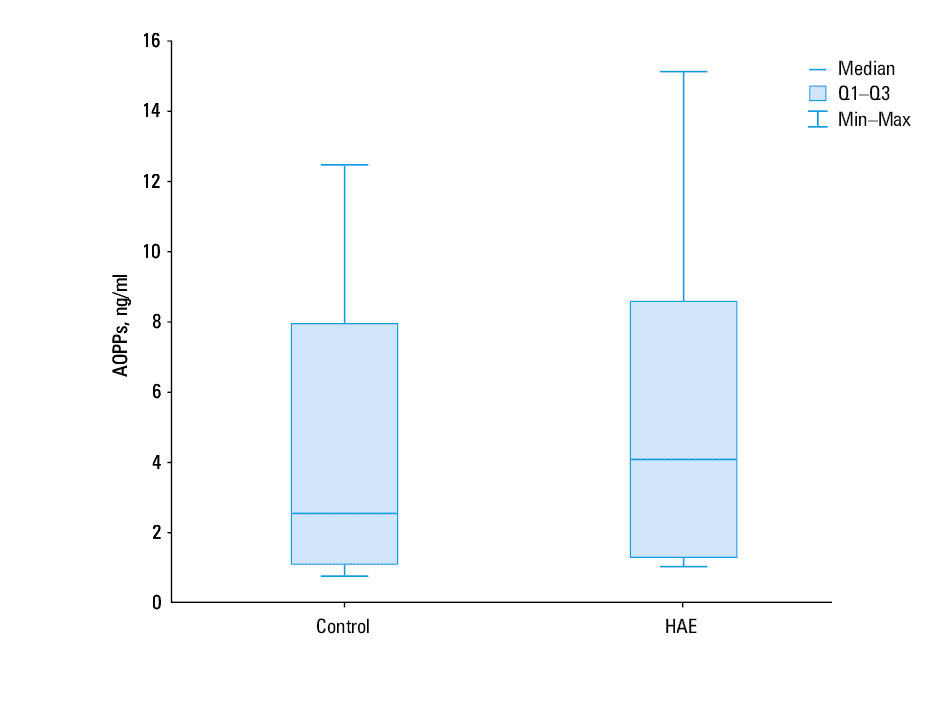
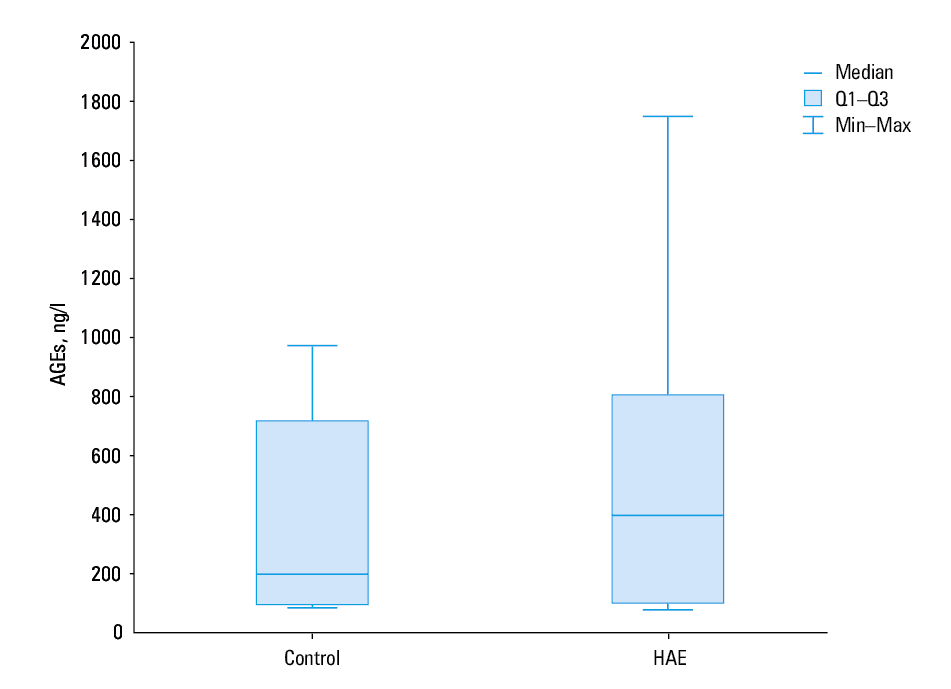
The median plasma levels of AOPPs and AGEs were similar in patient and control groups (P = 0.28 and P = 0.5, respectively; Figures 3 and 4). Increased AOPP levels in patients with HAE were positively correlated with increased AGE values (r = 0.93, P <0.00001). A similar correlation was also observed in controls (r = 0.93, P <0.00001).
In our study, exogenous bradykinin was shown to act as an antioxidant by lowering basal and H2O2-induced ROS levels in PBMCs. Exposure to bradykinin reduced basal ROS levels in patients with HAE (median [Q1; Q3], 2967 RFUs [634; 3800] vs 2617 RFUs [2119; 3051]; P = 0.03; Figure 5A), but not in controls (median [Q1; Q3], 2147 RFUs [1690; 2786] vs 2022 RFUs [1347; 2392]; P = 0.31; Figure 5B). Moreover, exposure to bradykinin decreased also H2O2-induced ROS levels in patients with HAE (median, 6672 RFUs [IQR, 5140–10 494] vs 5554 RFUs [IQR, 3854–6989]; P = 0.046; Figure 6A), but not in controls (median, 4352 RFUs [IQR, 3390–5531] vs 4126 RFUs [IQR, 2726–4486]; P = 0.23; Figure 6B).
Discussion
To our knowledge, our study is the first to show that patients with type I or II C1‑INH‑HAE have increased levels of basal ROS in PBMCs as compared with controls, which confirms that these patients have impaired redox balance with a significant prevalence of free radicals. Moreover, we found that the use of H2O2 as a nonspecific stimulus applied to PBMCs significantly increased ROS levels in patients with C1‑INH‑HAE when compared with controls. This indicates an increased tendency of PBMCs to produce free radicals when subjected to nonspecific stimuli and confirm disturbances in the redox homeostasis in this patient group.
In our study, we used DCFH‑DA to monitor changes in the redox status in PBMCs in response to the activation with an oxidative stimulus, because DCFH can be oxidized by various ROS. Thus, an increase of intracellular DCF fluorescence reflects the overall oxidative stress index in cells. Oxidative stress occurs in cells when the generation of ROS overwhelms the cells’ natural antioxidant defenses. DCFH‑DA may be used as a redox indicator probe that responds to changes in intracellular iron signaling or peroxynitrite formation.17
Moreover, in our study, we used AOPPs and AGEs, modified by interactions with ROS in the blood, as additional biomarkers of oxidative stress, which tends to be elevated in patients with C1‑INH‑HAE. However, the difference in AOPP and AGE levels between patients and controls was not significant. We observed that AGE levels higher than 1000 ng/l were present only in plasma samples from patients with C1‑INH‑HAE but not in healthy controls. Del Giacco et al18 studied patients with C1‑INH‑HAE as well as those with HAE with mutations in the F12 gene (FXII‑HAE). They reported that AGE levels were elevated only in the C1‑INH‑HAE group, as compared with controls, but not in the FXII‑HAE group. In contrast, C1‑INH deficiency did not seem to affect AOPP levels, as both C1‑INH‑HAE and FXII‑HAE groups showed elevated AOPP levels, as compared with controls.18 Therefore, further research on a larger group of patients and using the same measurement methods is needed to verify the values of these promising and important parameters for the assessment of oxidative stress involved in the pathophysiology of HAE.
Oxidative stress results from an imbalance between endogenous production of free ROS and inadequate effectiveness of antioxidant defense mechanisms. This imbalance can worsen inflammation and injury conditions by enhancing the release of proinflammatory cytokines and altering enzymatic function,10,19 by activating the complement20-22 and kinin systems as well as kinin receptors,23-27 by endothelial cell activation and dysfunction,25,28-31 as well as by affecting gene expression.32 C1‑inhibitor deficiency can aggravate these effects, especially by impaired redox homeostasis and increased oxidative stress in patients with C1‑INH‑HAE, as shown by Del Giacco et al18 and also by our team.
Our results concerning the effect of exogenous bradykinin on basal and H2O2-induced ROS levels in PBMCs in patients with C1‑INH‑HAE showed that bradykinin exerted a significant antioxidant effect in this group of patients. The lack of antioxidant bradykinin action in healthy controls may be associated with normal C1‑INH levels or may indicate that this effect manifests only in the case of increased oxidative stress.
To our knowledge, this is the first study confirming the antioxidant action of bradykinin in C1‑INH‑HAE. This seems to corroborate the findings reported so far in animal models33,34 as well as experimental studies on degenerative disease and cell aging processes in humans.24,25,35-39
Our results concerning oxidative stress in C1‑INH‑HAE as well as the effect of bradykinin on oxidative stress in PBMCs in this patient population were presented as preliminary findings during 2018 Bradykinin Symposium in Berlin.40 They are generally consistent with the recent study by Del Giacco et al,18 who indicated the role of oxidative stress in the pathophysiology of C1‑INH‑HAE.
We focused on the effect of bradykinin on PBMCs, as these cells can be separated from an easily accessible specimen; thus, we chose flow cytometry analysis with DCFH‑DA as a fluorescent probe. Flow cytometry is one of the most powerful tools for single‑cell analysis of the immune system used for many years to evaluate oxidative burst in numerous conditions, such as autoimmune neutropenia and asymptomatic HIV infection.41 DCFH‑DA has been also widely used for the detection of ROS and reactive nitrogen species, especially in mononuclear leukocytes (while other fluorescent probes are used only for polymorphonuclear leukocytes). This probe detects more reactive species (HO·, ONOO–, ROO·, NO2·, H2O2) than other probes. Isolation of PBMCs from whole blood and stabilization in the medium allowed us to avoid some factors interfering with ROS determination.42
Although the major effects of bradykinin are exerted on the endothelium, previous data showed the expression of B2R and B1R on the surface of PBMCs, indicating other target cells for bradykinin action.43 Although B1R is a potent activator of inducible nitric oxide and NADPH oxidase, potentially leading to oxidative stress, the role of B2R in this process is unclear. Based on a recent study on the role of bradykinin in neuroprotection, the molecular mechanisms of bradykinin’s action involve downregulation of the expression of caspase 1, interleukins 1β and 18, and cleaved gasdermin D (a key executioner of pyroptosis).44 Moreover, human tissue kallikrein gene delivery was found to protect against cerebral ischemia–reperfusion injury through B2R activation, and the proposed novel signaling mechanisms involved the activation of Homer1b/c‑ERK1/2 and Homer1b/c‑PI3K‑Akt signaling pathways.45 Based on our results, the next step in further research on the antioxidant activity of bradykinin in the model of PBMCs in patients with C1‑INH‑HAE should be a transcriptomics analysis of the signaling pathways.
Patients with C1‑INH‑HAE have significantly elevated intracellular ROS levels in PBMCs compared with healthy individuals. In the case of an adverse process such as oxidative stress, when redox balance is disturbed, different antioxidants may decrease ROS levels to some extent. Nevertheless, a number of free radicals in the cells of healthy controls is needed as signaling molecules, and ROS are generated continuously in physiological processes. Therefore, this so called “oxidative eustress” is not affected by antioxidants. It is known that cytokines produced by macrophages and mast cells, occurring in various diseases including HAE, promote overexpression of NADPH oxidase, which is the main source of excess ROS (apart from nitric oxide synthase and xanthine oxidase) in ischemia–reperfusion injury.18,23,34 Recently, it has been reported that in the state of oxidative stress mediated by hypoxia–reperfusion injury, the expression of B2R is significantly increased.44,45 This receptor may play a neuroprotective role in hypoxia–reoxygenation injury related to the pyroptosis pathway. It could be hypothesized that the same upregulation of the B2R expression occurs under the condition of oxidative stress documented in patients with C1‑INH‑HAE. However, further research is needed to fully explain the role of B1R and B2R as well as the endothelium and oxidative stress in the pathophysiology of C1‑INH‑HAE.
Although some previous studies have indicated the antioxidant property of bradykinin in animal models, the involvement of bradykinin in intracellular redox processes has not been fully elucidated. Bradykinin was found to protect endothelial cells against oxidative stress or enhanced ROS generation.29,31 It seems that the effect of bradykinin on the intracellular redox state depends on the type of the receptor with which the kinins or their derivatives interact.1,18,34
A study using bradykinin receptor–null mice indicated that nonselective stimulation of B1R and B2R is likely to suppress oxidative stress and diabetic nephropathy,23 while other studies suggested that the selective activation of B2R25 and inhibition of B1R could be beneficial.27,46 Data from studies on the vascular system of diabetic rats showed that B1R enhances superoxide anion radical formation by activating NADPH oxidase.18,27,46 Moreover, it has been shown that oxidative stress caused by elevated superoxide anion production induces expression of the kinin B1R in the liver and brain of diabetic rats.46-48
In light of recent findings, the cooperation between G protein–coupled receptors makes the signal transduction pathways of active peptides even more complex.18,49 Thus, oligomerization of bradykinin receptors and other receptors may modulate physiological processes and partially account for the discrepancies between bradykinin effects reported in different studies. Therefore, the expression and activation of B1R and B2R in patients with C1‑INH‑HAE should be investigated in further studies.
Bradykinin is known to induce nitric oxide synthase and release, which may lead to an increased production of peroxynitrite radicals, especially when antioxidant systems are not efficient. At the same time, bradykinin was proved to stimulate the activity of the antioxidant enzymes superoxide dismutase and catalase in endothelial cells.18,49 This recent evidence corroborates a previous study reporting an increase in superoxide dismutase, catalase, and glutathione peroxidase activity in hyperglycemic rats.47,48 On the basis of these findings as well as our data, a hypothesis may be proposed that one of the various bradykinin effects is to maintain the redox balance.
One of the limitations of our study is the use of a single measurement of bradykinin levels. The use of different concentrations of bradykinin in PBMC samples would be valuable, but it was not possible owing to the limited amount of PBMC specimen, as only a limited blood volume could be drawn from patients according to ethical standards. Thus, the concentration of bradykinin (100 pM) was chosen based on data on serum bradykinin concentrations during acute attacks of angioedema, provided in reliable literature sources.50 Our results should be confirmed in a larger study, together with the assessment of a dose dependency of bradykinin’s antioxidant action in C1‑INH‑HAE.34
In our study, exposure to bradykinin decreased also H2O2-induced ROS levels in patients with C1‑INH‑HAE. It seems that in these patients, oxidative stress disturbing redox balance may induce an increase in bradykinin levels in a feedback reaction in order to recover impaired redox homeostasis. Future research should focus on elucidating the role of kinins and kinin receptors as well as the endothelium in the pathomechanism of bradykinin‑mediated angioedema. A full explanation of the role of oxidative stress and antioxidant action of bradykinin might have important implications for the prevention and treatment of C1‑INH‑HAE.18,35,40
In conclusion, our study is the first to reveal a significant increase in basal and H2O2-induced ROS levels in patients with C1‑INH‑HAE as well as antioxidant action of exogenous bradykinin. It seems also that in patients with C1‑INH‑HAE, elevated oxidative stress may induce an increase of bradykinin levels as a natural antioxidant as a result of a feedback reaction provoking an angioedema attack. Further studies on the role of oxidative stress and antioxidant action of bradykinin might have important implications for the prevention and treatment of C1‑INH‑HAE.
- Caccia S, Suffritti C, Cicardi M. Pathophysiology of hereditary angioedema. Pediatr Allergy Immunol Pulmonol. 2014; 27: 159‑163. | Crossref
- Zuraw BL, Christiansen SC. HAE pathophysiology and underlying mechanisms. Clinic Rev Allerg Immunol. 2016; 51: 216‑229. | Crossref
- Kaplan AP, Joseph K. Pathogenesis of hereditary angioedema: the role of the bradykinin‑forming cascade. Immunol Allergy Clin North Am. 2017; 37: 513‑525. | Crossref
- Kaplan AP, Maas C. The search for biomarkers in hereditary angioedema. Front Med. 2017; 22: 206. | Crossref
- Obtułowicz K, Dyga W, Natorska J, et al. Elevated thrombin generation and factor VIII activity during angioedema attack in patients with hereditary C1 inhibitor deficiency. Pol Arch Intern Med. 2019; 129: 936‑938. | Crossref
ARTICLE INFORMATION