From hypertrophic cardiomyopathy to transthyretin amyloidosis: an unusual case and challenging diagnosis



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
From hypertrophic cardiomyopathy to transthyretin amyloidosis: an unusual case and challenging diagnosis
Transthyretin amyloidosis (ATTR) is a protein‑misfolding disease, in which amyloid fibrils infiltrate the myocardium.1 On echocardiography, ATTR may present as a phenocopy of hypertrophic cardiomyopathy (HCM). Several diagnostic tools may facilitate the diagnosis, such as echocardiography, scintigraphy, biomarkers, or genetic analysis.1 We report a case that highlights the role of cardiac biomarkers, extracardiac symptoms, and genetic analysis in the differential diagnosis of cardiac amyloidosis.
A 47‑year‑old man with heart failure (New York Heart Association class II) and suspicion of HCM or amyloidosis was referred to our hospital for further diagnostic workup. Doppler echocardiography revealed asymmetric left ventricular (LV) septal hypertrophy (2.2 cm during diastole; Figure 1A) with preserved LV ejection fraction (61%), and LV outflow tract gradient of 22 mm Hg at rest and 36 mm Hg after provocation. The ratio of early to late diastolic transmitral flow velocity (E/A) was 1.6, indicating a nonrestrictive filling pattern. The global longitudinal strain (GLS) was preserved in the apical segments, and there was no “apical sparing” pattern typical for ATTR. The mean GLS was –18.3%, but there was a marked abnormal gradient of deformation between the basic and apical segments (minimum value, –4%; maximum value, –32%; Figure 1B). For comparison, Figure 1C shows a totally abnormal GLS pattern of a patient with more advanced ATTR.2
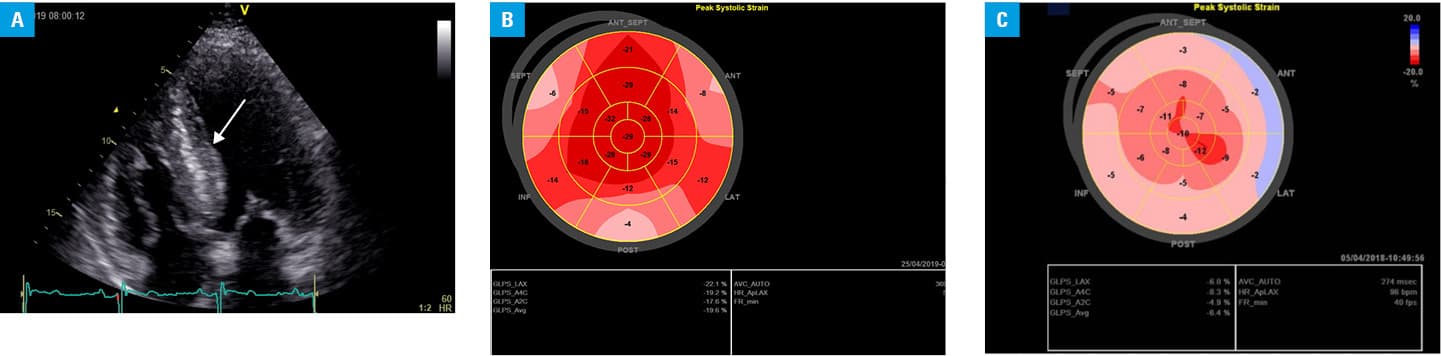
The patient had normal high‑sensitive troponin I (hs‑TnI, 9.3 ng/ml) and N‑terminal fragment of the prohormone brain natriuretic peptide (NT‑proBNP, 98.5 pg/ml) levels (double‑negative biomarkers), which might be explained by an early stage of the disease. A year later, the NT‑proBNP level was elevated (374 pg/ml; reference range, 125 pg/ml). Next, the patient experienced new‑onset hand numbness (primarily in the left hand), indicating carpal tunnel syndrome with neural compression. This led us to change the tentative diagnosis from HCM to cardiac amyloidosis.1,2 We performed 99mTc‑3, 3‑diphosphono‑1, 2‑propanodicarboxylic acid scintigraphy, which showed radiotracer uptake in the heart, typical of ATTR.1,2
Genetic analysis revealed the ATTR variant c.325G>A (p.Glu109Lys). This variant E109K in the TTR gene cannot be found in the ClinVar database; however, 2 other substitutions in the position of 109 in the TTR gene have been reported as pathogenic (E109Q) or of uncertain significance (E109D). This finding supports the diagnosis of ATTR.
The GLS polar plot map did not provide any conclusive findings. However, scintigraphy yielded strong evidence supporting the diagnosis of ATTP confirmed by genotyping. The patient presented with normal hs‑TnI and NT‑proBNP levels despite several echocardiographic features typical of HCM with mild obstruction. Therefore, we postulate that the normal levels of hs‑TnI and NT‑proBNP may help distinguish HCM from ATTR (at an early stage of the disease).
Our hypothesis could be useful in interpreting the study by Zhang et al.3 The authors did not provide data on the minimal value of NT‑proBNP in the HCM subgroup with negative troponin results, but observed very high median and lower quartile NT‑proBNP values (median [IQR], 1166.7 [775.5–1818.5] pg/ml). Probably, there were no troponin/NT‑proBNP–negative patients. In the Methods section, the authors did not mention that the HCM phenocopies were excluded. They did not use any specific tests or examinations for a differential diagnosis of ATTR. It might be speculated, in our opinion, that some patients with HCM and normal NT‑proBNP levels had undiagnosed ATTR.
- Agha AM, Parwani P, Guha A, et al. Role of cardiovascular imaging for the diagnosis and prognosis of cardiac amyloidosis. Open Heart. 2018; 5: e000881. | Crossref
- Rajtar‑Salwa R, Gębka A, Petkow‑Dimitrow P. Non‑invasive cardiac imaging methods in transthyretin amyloidosis. Kardiol Pol. 2019; 77: 234. | Crossref
- Zhang C, Liu R, Yuan J, et al. Significance and determinants of cardiac troponin I in patients with obstructive hypertrophic cardiomyopathy. Am J Cardiol. 2015; 116: 1744‑1755. | Crossref
ARTICLE INFORMATION