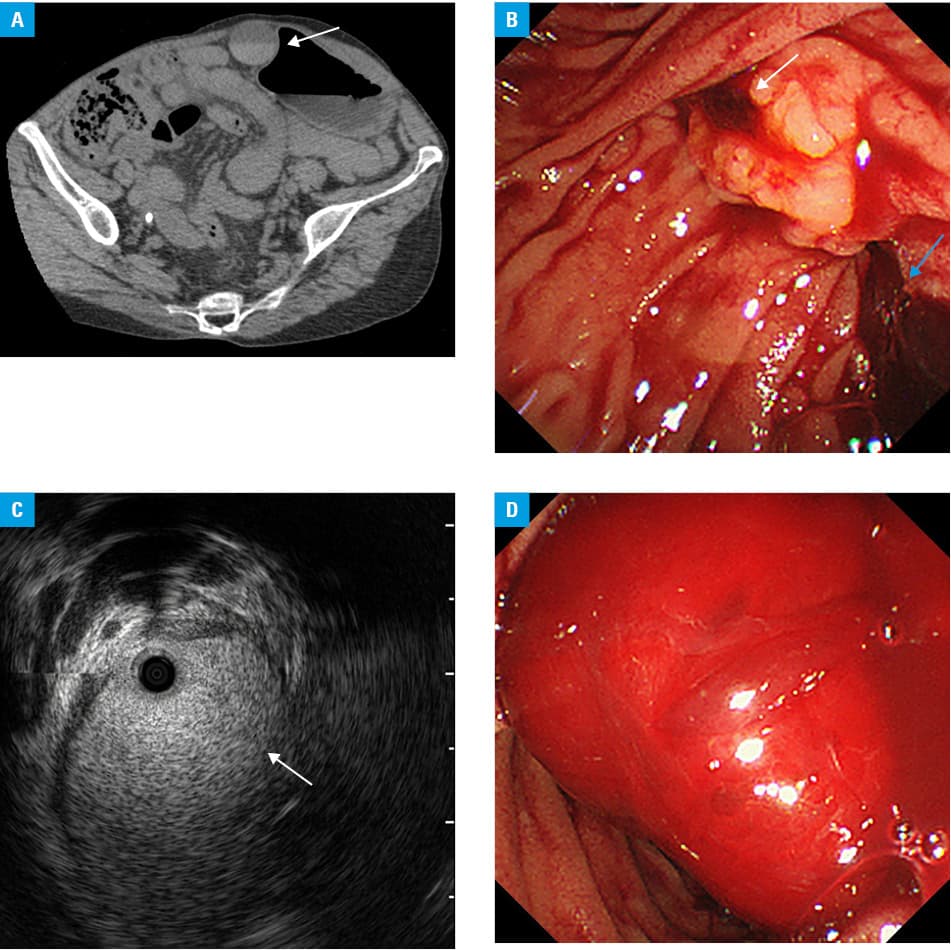
A 54‑year‑old woman presented with postprandial right upper quadrant pain. Her medical history included type 2 respiratory failure due to severe congenital scoliosis, home mechanical ventilation with tracheotomy, right chronic empyema, complicated urinary tract infection, and hereditary hemorrhagic telangiectasia (HHT). Physical examination demonstrated stable vital signs and positive Murphy sign. Laboratory tests showed white blood cell count of 8500/mm3, hemoglobin level of 7.2 g/dl, C‑reactive protein level of 2.15 mg/dl (reference range <0.14 mg/dl), total bilirubin level of 0.7 mg/dl (reference range, 0.4–1.5 mg/dl), aspartate aminotransferase level of 44 U/l (reference range, 13–30 U/l), alanine aminotransferase level of 64 U/l (reference range, 7–23 U/l), γ-glutamyl transpeptidase level of 102 U/l (reference range, 9–32 U/l), alkaline phosphatase level of 751 U/l (reference range, 106–322 U/l), and amylase of level 146 U/l (reference range, 44–132 U/l). As she had allergy for intravenous iodinated contrast material, plain computed tomography scan was performed, showing high‑density fluid in the gall bladder, which was suggestive of blood (Figure 1A). With a suspicion of acute cholangitis, endoscopic retrograde cholangiopancreatography was performed, disclosing active hemobilia (Figure 1B). Intraductal ultrasonography showed highly echogenic material in the dilated common bile duct, which was suggestive of blood (Figure 1C). Although the presence of apparent hepatic or biliary telangiectasias and arteriovenous malformations (AVMs) could not be proved, acute cholangitis by hemobilia associated with HHT was clinically diagnosed. No lithiasis was present and a biliary plastic stent was placed, resulting in clinical improvement. The patient’s symptoms recurred; however, endoscopic retrograde cholangiopancreatography was repeated 2 weeks later. After the stent impacted with clot was removed, massive hemobilia was drained and no further intervention was added (Figure 1D). She experienced recurrent hemobilia and spontaneous improvement until her death by respiratory failure 2 years later.
Hereditary hemorrhagic telangiectasia, also known as Rendu–Osler–Weber syndrome, is a disorder inherited as an autosomal dominant trait, characterized by epistaxis, mucocutaneous telangiectasias, and visceral AVMs, which lead to chronic hemorrhage and anemia. Symptomatic hepatic and biliary AVMs occur very rarely. Hepatic involvement may result in high‑output heart failure, portal hypertension due to hepatic artery to portal vein shunt, hepatomegaly, and jaundice, whereas biliary diseases include cholestasis, recurrent cholangitis, and hepatic disintegration.1 Most cases still have difficulties in diagnosis of apparent bleeding points. Moreover, no strong correlation was observed between computed tomography findings and the clinical subtypes of hepatobiliary lesions in HHT.2
Although AVMs could not be proved inside the biliary tract in this case, fortunately hemobilia was followed conservatively. In cases of telangiectasias detected by angiography or endoscopy including cholangioscopy, endovascular intervention, and endoscopic thermal ablation can be effective for hemostasis.3,4 The present therapy of the disease is intended to reduce the symptoms. Although mechanism‑based therapy is not available so far, recent progress has been made using drugs that target vascular endothelial growth factor and the angiogenic pathway with the use of the humanized monoclonal antibody against vascular endothelial growth factor (bevacizumab).1,5 In conclusion, although rare, hemobilia should be included in the differential diagnosis of gastrointestinal bleeding in HHT.
- Sabbà C, Pompili M. Review article: the hepatic manifestations of hereditary haemorrhagic telangiectasia. Aliment Pharmacol Ther. 2008; 28: 523‑533. | Crossref
- Wu JS, Saluja S, Garcia‑Tsao G, et al. Liver involvement in hereditary hemorrhagic telangiectasia: CT and clinical findings do not correlate in symptomatic patients. AJR Am J Roentgenol. 2006; 187: W399‑W405. | Crossref
- Hayashi S, Baba Y, Ueno K, et al. Small arteriovenous malformation of the common bile duct causing hemobilia in a patient with hereditary hemorrhagic telangiectasia. Cardiovasc Intervent Radiol. 2008; 31 (suppl 2): S131‑S134. | Crossref
- Komaki Y, Kanmura S, Funakawa K, et al. A case of hereditary hemorrhagic telangiectasia with repeated hemobilia arrested by argon plasma coagulation under direct peroral cholangioscopy. Gastrointest Endosc. 2014; 80: 528‑529. | Crossref
- Robert F, Desroches‑Castan A, Bailly S, et al. Future treatments for hereditary hemorrhagic telangiectasia. Orphanet J Rare Dis. 2020; 15: 4. | Crossref
ARTICLE INFORMATION