A 64‑year‑old woman with arterial hypertension and a history of drug and alcohol abuse was referred to our hospital because of painless jaundice lasting for 8 months. Initial examination revealed significantly increased levels of aspartate transaminase (AST), 1391 U/l (reference range, 0–31 U/l); alanine transaminase (ALT), 971 U/l (reference range, 0–35 U/l); gamma‑glutamyltransferase (GGTP), 163 U/l (reference range, 0–35 U/l); and bilirubin, 15.2 mg/dl (reference range, 0.2–1.2 mg/dl). Viral hepatitis was excluded. Antinuclear (titer, 1:100) and anti–smooth muscle antibodies (titer, 1:100) were detected, but no liver‑kidney antimicrosomal type 1, anti–soluble liver antigen and liver‑pancreas and antimitochondrial antibodies were found. However, serum immunoglobulin G (IgG) levels were elevated at 41.17 g/l (reference range, 7–16 g/l) and its subtype G4 (IgG4) level was also significantly higher at 4.83 g/l (reference range, 0.03–2.01 g/l). Biopsy of the papilla of Vater also revealed IgG4‑positive (IgG4+) cells. Liver biopsy indicated portal inflammation, interface and lobular hepatitis, plasma cell infiltration, and rosette formation (Figure 1A–1C), whereas no bile duct damage was noted. Findings from the liver biopsy were still inconclusive. The constellation of morphological features did not meet explicit diagnostic criteria for any specific liver disease, among others, for the classical type of autoimmune hepatitis (AIH). Histological examination suggested IgG4‑related disease. However, the quantitative criteria of the current diagnostic consensus1 were not met when immunohistochemical examination and quantitative analysis of IgG and IgG4 were considered. Four IgG4+ cells per high‑power field (HPF) were identified (Figure 1D). Additionally, magnetic resonance imaging and endoscopic retrograde cholangiopancreatography excluded biliary tract pathology. Nevertheless, the patient met the criteria for AIH according to the International Autoimmune Hepatitis Group, scoring 7 points (antinuclear or anti–smooth muscle antibodies ≥1:8, IgG level more than 1.1‑fold higher than the upper limit of normal, liver histology compatible with AIH, and absence of viral hepatitis).2 Prednisone at a dose of 40 mg/d was initiated and resulted in a significant decrease of liver enzyme activity (AST, 41 U/l; ALT, 24 U/l; GGTP, 56 U/l), bilirubin (2.4 mg/dl), and IgG (16.38 g/l) levels at 8‑month follow‑up.
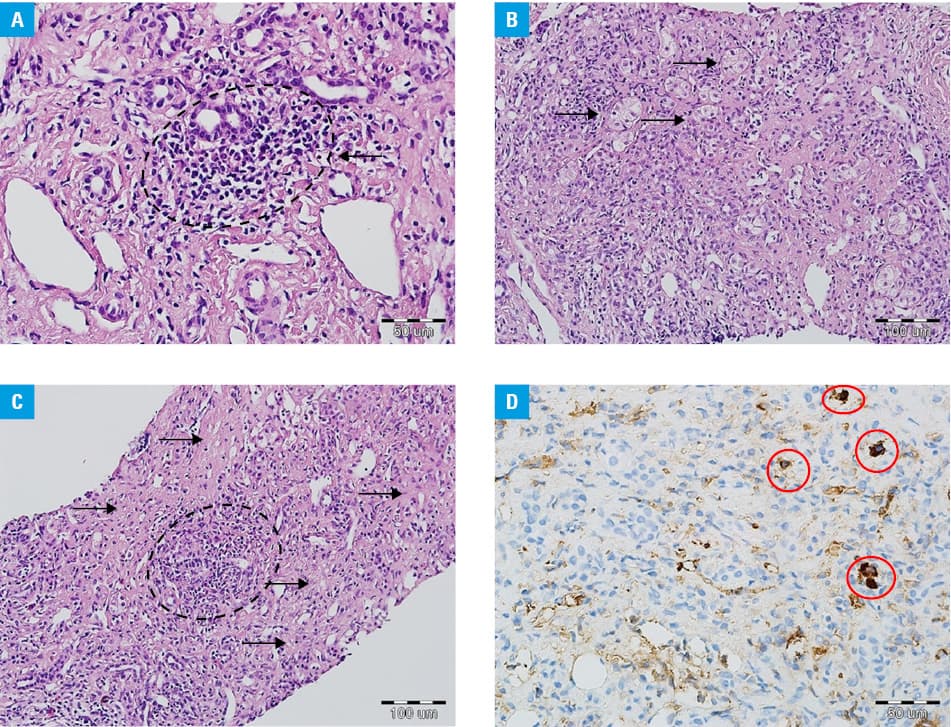
Immunoglobulin G4–related disease is a systemic disease characterized by an increased concentration of IgG4 in serum and IgG4+ cell infiltrations in multiple organs.3 Recently, there has been a discussion as to whether this abnormality is a hepatic manifestation of systemic IgG4‑related disease (IgG4‑related hepatopathy) or a subtype of AIH (IgG4‑related AIH).4 Minaga et al5 summarized the clinicopathological features of IgG4‑related AIH, identifying this condition based on hepatic accumulation of IgG4+ cells in patients with AIH. A number of more than 5 IgG4+ cells per HPF was regarded as a significant accumulation of IgG‑expressing plasma cells in the liver. However, in our patient, we found only 4 IgG4+ cells per HPF. Our limitation was the HPF applied ( × 50), as it was lower ( × 40) in the study by Minaga et al,5 which means that we assessed a smaller part with a minor biopsy core of the liver and it could impact the analysis.
Here, we reported a rare and atypical case of AIH, accompanied by a marked elevation of serum IgG4 concentration and inconclusive histological findings, which met the quality criteria for IgG4‑related disease.
- Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4‑related disease. Mod Pathol. 2012; 25: 1181‑1192.
- Hennes EM, Zeniya M, Czaja AJ, et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology. 2008; 48: 169‑176. | Crossref
- Okazaki K, Uchida K. Current perspectives on autoimmune pancreatitis and IgG4‑related disease. Proc Jpn Acad Ser B Phys Biol Sci. 2018; 94: 412‑427. | Crossref
- Joshi D, Webster GJ. Biliary and hepatic involvement in IgG4‑related disease. Aliment Pharmacol Ther. 2014; 40: 1251‑1261. | Crossref
- Minaga K, Watanabe T, Chung H, Kudo M. Autoimmune hepatitis and IgG4‑related disease. World J Gastroenterol. 2019; 25: 2308‑2314. | Crossref
ARTICLE INFORMATION