Intrafamilial variability of cardiovascular abnormalities associated with the p.R460H mutation of the TGFBR2 gene



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Intrafamilial variability of cardiovascular abnormalities associated with the p.R460H mutation of the TGFBR2 gene
The p.R460H mutation of the transforming growth factor β receptor 2 gene (TGFBR2) has been reported in Loeys–Dietz syndrome,1 familial thoracic aortic aneurysms and dissection,2,3 and in a patient meeting the revised Ghent criteria for the diagnosis of Marfan syndrome.4,5
We clinically examined 3 generations of a family with a wide spectrum of cardiovascular abnormalities and performed a genetic examination to identify the common cause of the disease.
Based on next‑generation sequencing, a pathogenic p.R460H variant in the TGFBR2 gene was identified in the affected patient. Further Sanger sequencing confirmed the p.R460H mutation in 8 subjects in 3 generations.
All individuals underwent transthoracic echocardiography. Magnetic resonance angiography of the arterial system was performed in 1 patient, and computed tomography angiography of the aorta in 7 patients, of the carotid and intracranial arteries in 2, and of the coronary arteries in other 2.
The abnormalities of the vascular system were identified in all carriers (Figure 1A–1F; details are shown in Supplementary material, Table S1). Among 8 patients, the most prevalent manifestation was aortic root aneurysm (in 6 individuals) and, surprisingly, atrial septal defect (in 4 individuals). Both ascending aortic aneurysm and descending aortic dilatation were found in 3 patients. Vascular complications were noted in 3 persons (spontaneous dissection of both coronary arteries and 2 type A thoracic aortic dissections). In the family, there were 4 sudden deaths under 40 years of age due to unknown cause: 1 in a carrier with aortic root aneurysm who was eligible for surgery (subject III 9), another 1 in an obligate carrier (II 2), and 2 in subjects of unknown genetic status (III 5 and IV 8) (Figure 1G). Hypertension was reported in 4 individuals (50%). Three patients underwent valve‑sparing root replacement with the aortic root dimensions of 50 mm, 47 mm, and 47 mm (David procedure), and another patient had surgery due to abdominal aortic aneurysm.
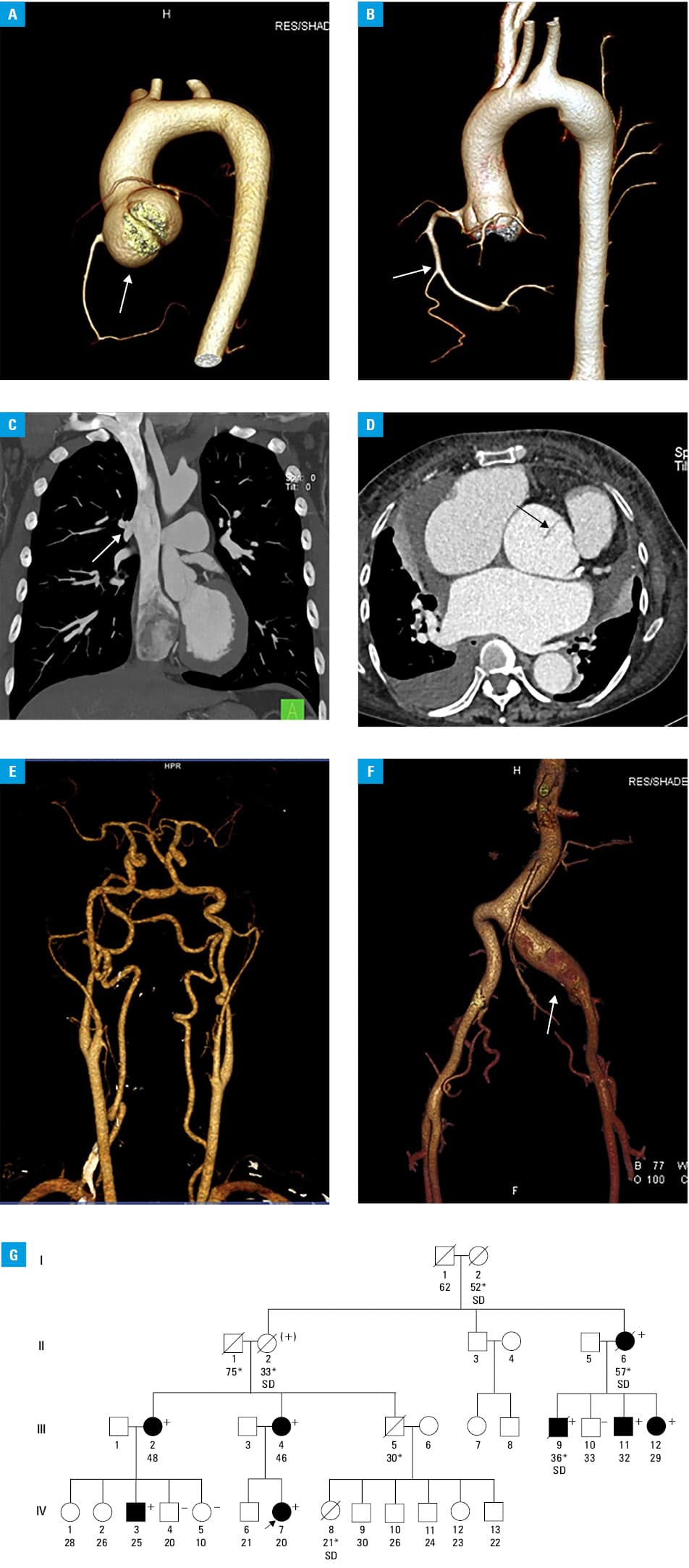
Involvement of other systems was common: skeletal findings (pectus deformity, scoliosis, arachnodactyly, joint laxity, and clubfoot) were present in 6 patients, craniofacial abnormalities in 4 (hypertelorism, dolichocephaly, cleft palate, and blue sclerae), and cutaneous manifestations in 4 (skin striae, easy bruising, and vitiligo); 2 subjects had inguinal hernias.
Loeys–Dietz syndrome is clinically characterized by thoracic aortic aneurysms and dissections, generalized arterial tortuosity, aneurysms with dissections throughout the remaining arterial tree, congenital cardiac disease, and common multiple system signs.1 The p.R460H mutation of the TGFBR2 gene is located in the serine/threonine kinase domain of TGFBR2 and belongs to the hot‑spot mutations of the gene. Functional studies reported that p.R460H exhibited a dominant negative effect on extracellular signal‑regulated kinase and downstream signal transducers, leading to defective transforming growth factor β signaling.5 High morbidity and premature mortality, as well as a strikingly variable cardiovascular phenotype were documented in the presented family. Our family case shows that thoracic aortic aneurysm, both ascending and descending, accompanied by a variety of vasculopathies and congenital heart defects, is the most common cardiovascular abnormality associated with the p.R460H mutation of the TGFBR2 gene.
- Loeys BL, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF‑beta receptor. N Engl J Med. 2006; 355: 788‑798. | Crossref
- Pannu H, Fadulu VT, Chang J, et al. Mutations in transforming growth factor‑beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation. 2005; 112: 513‑520. | Crossref
- Law C, Bunyan D, Castle B, et al. Clinical features in a family with an R460H mutation in transforming growth factor beta receptor 2 gene. J Med Genet. 2006; 43: 908‑916. | Crossref
- Disabella E, Grasso M, Marziliano N, et al. Two novel and one known mutation of the TGFBR2 gene in Marfan syndrome not associated with FBN1 gene defects. Eur J Hum Genet. 2006; 14: 34‑38. | Crossref
- Horbelt D, Guo G, Robinson PN, et al. Quantitative analysis of TGFBR2 mutations in Marfan‑syndrome‑related disorders suggests a correlation between phenotypic severity and Smad signaling activity. J Cell Sci. 2010; 123: 4340‑4350. | Crossref
ARTICLE INFORMATION