Antithrombin deficiency as a still underdiagnosed thrombophilia: a primer for internists
Key words: antithrombin deficiency, inherited thrombophilia, molecular analysis, thrombophilia testing, venous thromboembolism



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Antithrombin deficiency as a still underdiagnosed thrombophilia: a primer for internists
Antithrombin is a key endogenous anticoagulant that also plays other roles in inflammation, immunity, and other processes. Congenital antithrombin deficiency is the most severe type of thrombophilia, yet characterized by a remarkable clinical heterogeneity. Here, as a primer for internists, we present a practical review of data regarding this disorder, focused on its molecular basis, diagnostic procedures, prognostic implications, and clinical management of patients suffering from this severe, and probably underdiagnosed, type of thrombophilia.
Introduction
The hemostatic system represents a delicate equilibrium between procoagulant and anticoagulant elements. Physiologically, 3 main anticoagulant pathways dominate in this equilibrium and enable blood flow through the vascular system: tissue factor inhibitor, protein C, and antithrombin pathways. Any distortion in the function of these anticoagulants will definitively increase the risk of a thrombotic event through an inefficient control of procoagulant reactions. This review focuses on probably the main endogenous anticoagulant, antithrombin, and the clinical consequences of its deficiency. The aim of the present article is to describe this protein and the disorder as well as to provide useful guidance for its proper diagnosis and management.
Antithrombin molecule
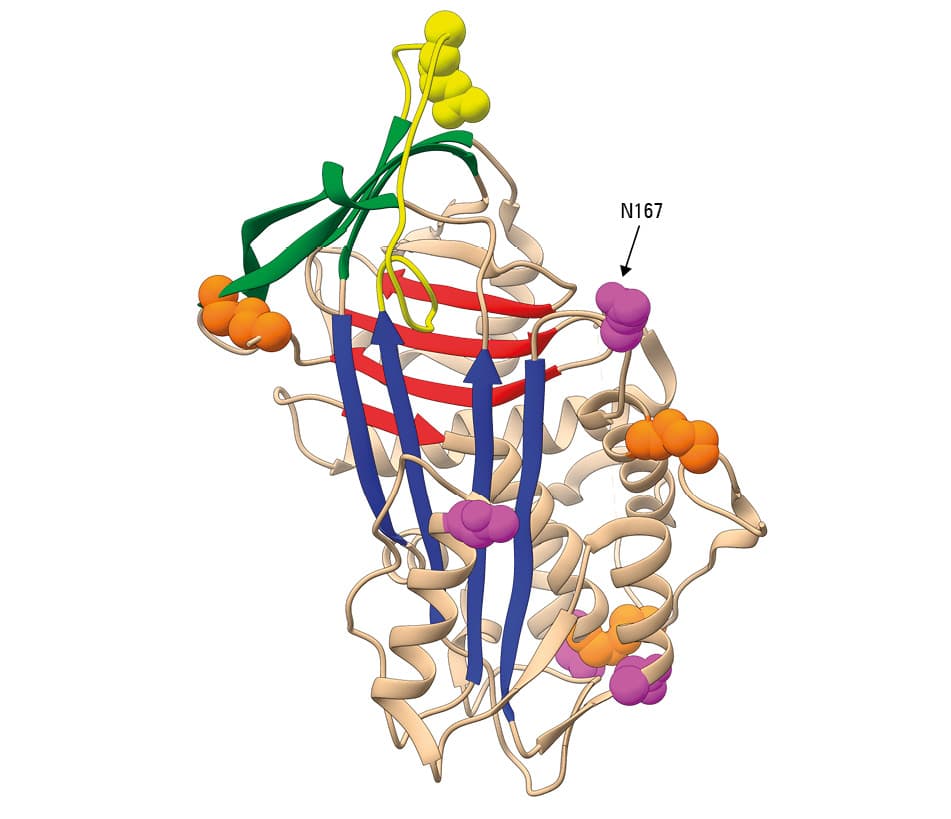
Antithrombin is a glycoprotein, a product of hepatic synthesis. The signal peptide of 32 amino acids guides the nascent protein to the endoplasmic reticulum, where the mature protein of 432 amino acids is subjected to 2 post‑translational modifications: N‑glycosylation at 4 asparagines (Asn128, Asn167, Asn187, and Asn224) and 3 cysteine disulfide bonds (Cys40–Cys160, Cys53–Cys127, and Cys279–Cys462) (figure 1). Since Asn167 is inefficiently glycosylated, there are 2 physiological glycoforms of antithrombin in plasma: α with 4 N‑glycans and a molecular weight of 58 kDa, which accounts for 90% of plasma antithrombin, and β (10% of plasma antithrombin) with 3 N‑glycans and a molecular weight of 56 kDa.1
- McCoy AJ, Pei XY, Skinner R, et al. Structure of beta‑antithrombin and the effect of glycosylation on antithrombin’s heparin affinity and activity. J Mol Biol. 2003; 326: 823‑833. | Crossref
- Huntington JA, Read RJ, Carrell RW. Structure of a serpin‑protease complex shows inhibition by deformation. Nature [Internet]. 2000; 407: 923‑926. | Crossref
- Corral J, Rivera J, Guerrero JA, et al. Latent and polymeric antithrombin: clearance and potential thrombotic risk. Exp Biol Med (Maywood). 2007; 232: 219‑226.
- van Boven HH, Lane DA. Antithrombin and its inherited deficiency states. Semin Hematol. 1997; 34: 188‑204.
- Björk I, Olson ST. Antithrombin. A bloody important serpin. Adv Exp Med Biol. 1997; 425: 17‑33.
ARTICLE INFORMATION