A 35‑year‑old woman with no family history of malignancy was hospitalized due to unintentional weight loss of 6 kg within 3 weeks, progressive weakness, and fever of unknown origin. Two weeks before the admission, she palpated a tumor in the left breast.
On examination, she was in good condition, with tumors in both breasts and tenderness of the pelvic region. Laboratory workup showed a slightly elevated serum C‑reactive protein level (28 mg/l; upper limit of normal, 5 mg/l), creatinine (1.5 mg/dl; reference range, 0.51–0.95 mg/dl), and D‑dimer (1149 ng/ml; upper limit of normal, 500 ng/ml). Sonography revealed nodular, solid‑cystic structures sized 79 × 58 mm and 93 × 72 mm in the right and left ovary, respectively. Mammography and ultrasonography of the breasts showed findings typical of category 4 Breast Imaging‑Reporting and Data System (BI‑RADS) (Figure 1A and 1B); the breast tumors were biopsied. Magnetic resonance imaging (MRI) of the pelvis revealed focal lesions of both ovaries (Figure 1C and 1D), numerous infiltrative foci within the bone structures, roots of the sacral spinal nerve 1 on the left side and atypical mass, which modelled both iliac veins (Figure 1E and 1F).
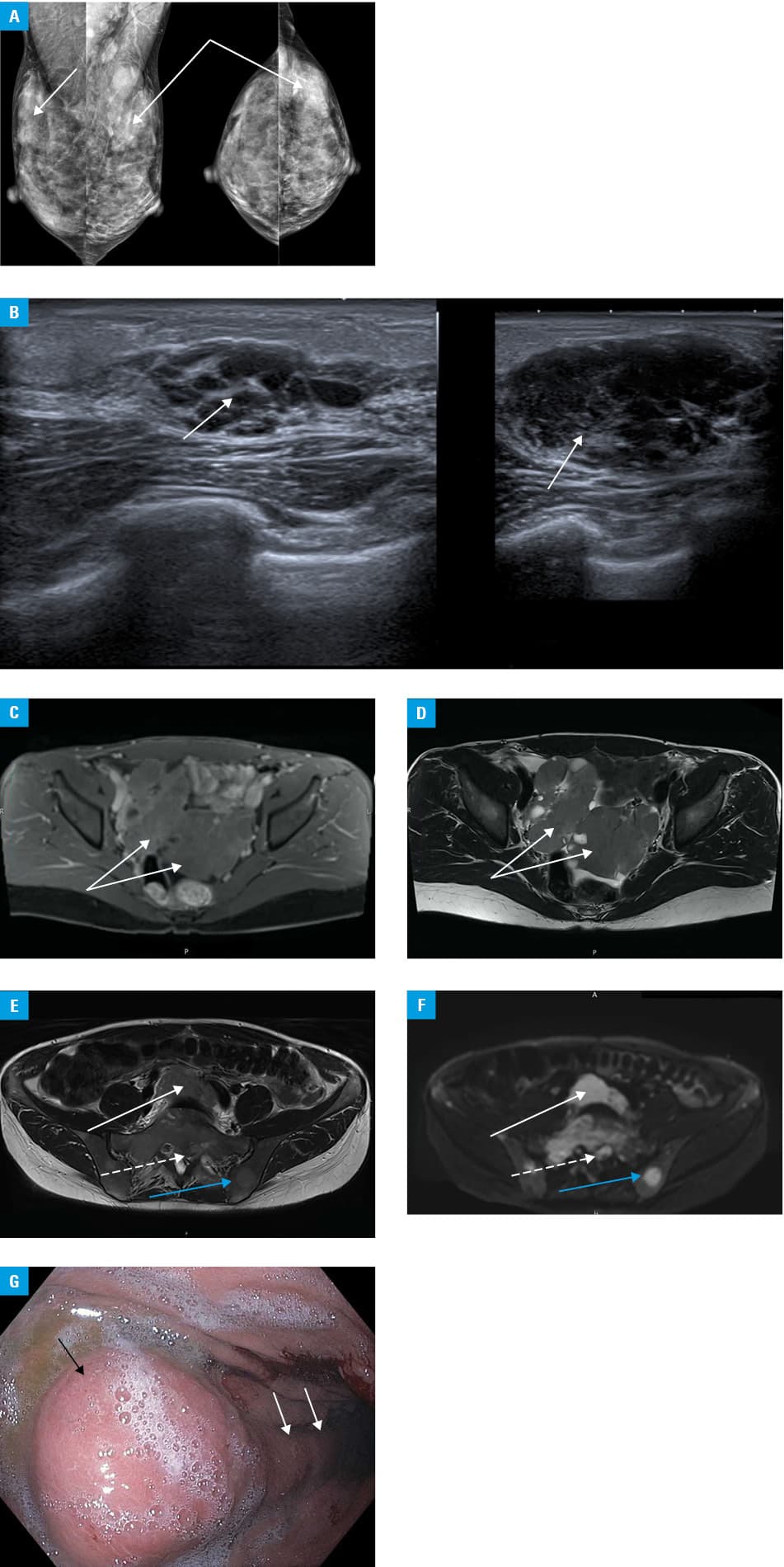
Computed tomography of the chest and abdominal cavity did not reveal any significant abnormalities, except for uncharacteristic image of the stomach wall, which prompted an upper gastrointestinal tract examination. Gastroscopy showed a 30‑mm tumor on the greater curvature of the stomach, covered with the congested mucosa and an infiltrated fold, extending towards the antral part of the stomach, which were biopsied (Figure 1G).
While the clinical presentation resembled a disseminated malignant neoplasm of unknown origin, we first suspected a breast‑ovary syndrome. Surprisingly, histologic examination showed no features of breast or ovarian cancer, but was suggestive of lymphoid tissue. Immunochemistry assays revealed the Burkitt lymphoma (BL) phenotype, with CD20(+), bcl‑6(+), CD10(+), bcl‑2(–), CD30(–), CD45(+), CD38(+), overexpression of c‑myc(+), and high Ki67 proliferation index (nearly 100%).
Burkitt lymphoma is a highly aggressive B cell non‑Hodgkin lymphoma, which derives from a mature germinal or postgerminal B cell, often presenting as a rapidly growing tumor with a very short doubling time, between 24 to 48 hours.1 It represents less than 5% of lymphoma cases in adults with the overall survival rate exceeding 70%.2,3 It disseminates quickly to extranodal sites, mainly the central nervous system, bone marrow, gut, head, and neck. Approximately 70% of newly diagnosed patients demonstrate advanced stages (III or IV) of the disease.4 Three types of BL have been identified, including endemic (50%–90%), immunodeficiency‑related (25%–40%), and sporadic (1%–2%).4 The sporadic one, most likely found in our patient, is predominantly diagnosed in Western Europe. It affects mostly younger patients, with a peak incidence at the ages of 11 years and 30 years (3.5 times more common in men);5 the abdomen is the most common site of sporadic BL, seen in 60% to 80% of patients.
In this case, BL developed simultaneously in the breast, stomach, and ovary. To the best of our knowledge, this is probably the first case of BL in this localization. We suggest that BL, although uncommon, should be considered in differential diagnosis in patients with malignancy of unknown origin and atypical course. Fortunately, overall survival in patients diagnosed with BL and aged less than 40 is significantly better than in the older ones.
- Casulo C, Friedberg J. Burkitt lymphoma – a rare but challenging lymphoma. Best Pract Res Clin Haematol. 2018; 31: 279‑284. | Crossref
- Bishop P, Rao V, Wilson W. Burkitt’s lymphoma: molecular pathogenesis and treatment. Cancer Investig. 2000; 18: 574‑583. | Crossref
- Ribrag V, Koscielny S, Bodaq J, et al. Rituximab and dose‑dense chemotherapy for adults with Burkitt’s lymphoma: a randomised, controlled, open‑label, phase 3 trial. Lancet. 2016; 387: 2402‑2411. | Crossref
- Jaffe E. The 2008 WHO classification of lymphomas: implications for clinical practice and translational research. Hematology Am Soc Hematol Educ Program. 2009; 523‑531. | Crossref
- Molyneux E, Rochford R, Griffin B, et al. Burkitt’s lymphoma. Lancet. 2012; 379: 1234‑1244. | Crossref
ARTICLE INFORMATION