A 51‑year‑old man with polycystic liver disease (PLD), diagnosed at the age of 48 years, was admitted to the Department of Gastroenterology and Hepatology because of increasing abdominal pain.
In the following year, he was referred to an outpatient gastroenterology clinic owing to increased abdominal girth, recurrent pain in the right upper abdomen and right flank, abdominal discomfort, early satiety, and dyspnea, especially at night. Due to reported complaints, he underwent percutaneous drainage of liver cysts 5 times. However, only partial short‑term symptom relief was achieved.
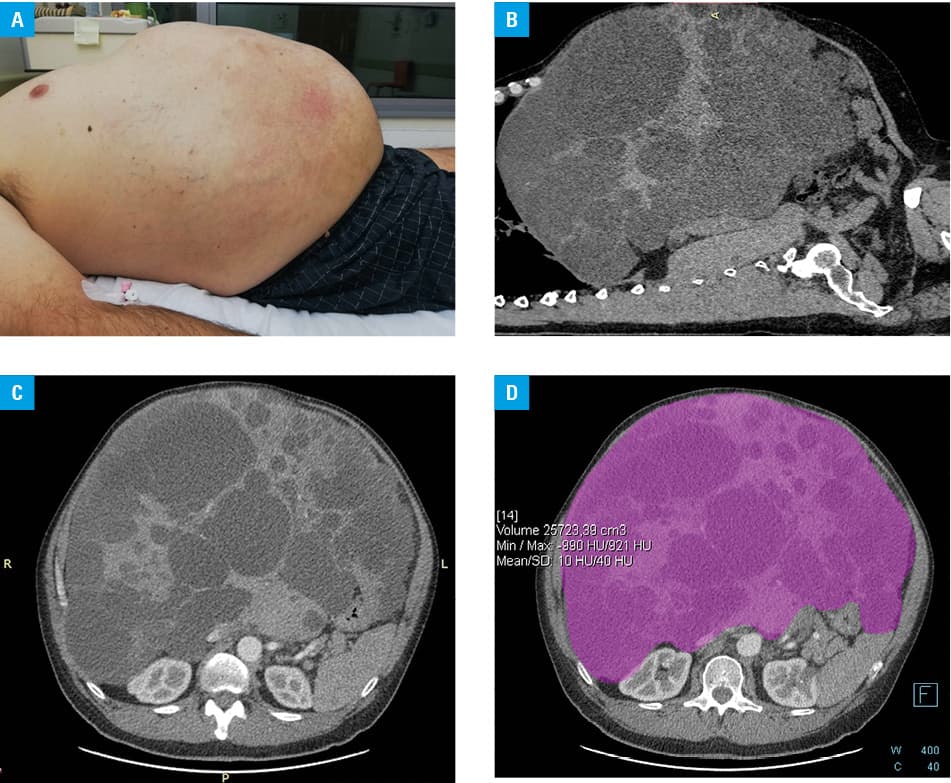
Clinical examination showed an enlarged liver, reaching into the right pelvic region, crossing the midline of the abdomen, and resulting in abdominal bulging (figure 1A). The anthropometric measurements were as follows: body weight, 127 kg; height, 191 cm; waist circumference, 140 cm; and body mass index, 34.8 kg/m2.
Laboratory tests demonstrated increased levels of total bilirubin (2 mg/dl; reference range <1.2 mg/dl), aspartate aminotransferase (133 U/l; reference range <34 U/l), alanine aminotransferase (168 U/l; reference range <55 U/l), γ-glutamyltransferase (2507 U/l; reference range <36 U/l), and alkaline phosphatase (664 U/l; reference range <100 U/l). Albumin, total protein, creatinine, carbohydrate antigen 19.9, and α-fetoprotein levels were within the reference range.
Multiphase contrast‑enhanced computed tomography was performed and confirmed hepatomegaly (craniocaudal dimension of 43 cm) with multiple Gigot type III liver cysts (figure 1B and 1C). The liver volume, calculated using a dedicated, commercially available software, was 25.7 l (figure 1D). The diameter of thin‑walled liver cysts varied between less than 1 cm and 16 cm and there were no signs of infection or hemorrhage. Also, no signs of compression of the main biliary ducts, hepatic venous outflow obstruction, or portal vein occlusion were found. Two cysts were seen in the right kidney and 3 in the left kidney.
Having evaluated the patient’s clinical status and therapeutic options, the man was found eligible for orthotopic liver transplant. The procedure was performed in February 2020.
Polycystic liver disease denotes a set of rare, genetic human disorders resulting from developmental alterations within the biliary tree.1 It is arbitrarily diagnosed when there are more than 20 fluid‑filled cysts in the liver.2 The condition is associated with 2 genetically distinct diseases: the primary phenotype in isolated polycystic liver disease and the extrarenal manifestation in autosomal dominant polycystic kidney disease.1-31‑3 The natural history of PLD is characterized by an increase in the number and volume of liver cysts.3 A total of 80% of patients remain asymptomatic, are usually diagnosed incidentally, and do not require any intervention.3 In 20% of patients, hepatomegaly can cause pain or compression of the surrounding organs, the diaphragm, or vasculature.3 Furthermore, symptoms can be induced by complications associated with liver cysts, such as: infections, hemorrhage, and rupture.1,3 In symptomatic patients, the main goal is to relieve symptoms and improve the quality of life.2 The current treatment options include: pharmacological treatment (somatostatin analogues), aspiration sclerotherapy, open or laparoscopic cyst fenestration with or without hepatic resection, and orthotopic liver transplant.4 Only a small group of selected patients with PLD require orthotopic liver transplant.1-4
- Cnossen WR, Drenth JPH. Polycystic liver disease: an overview of pathogenesis, clinical manifestations and management. Orphanet J Rare Dis. 2014; 9: 1‑13. | Crossref
- van Aerts RMM, van de Laarschot LFM, Banales JM, Drenth JPH. Clinical management of polycystic liver disease. J Hepatol. 2018; 68: 827‑837. | Crossref
- Abu‑Wasel B, Walsh C, Keough V, Molinari M. Pathophysiology, epidemiology, classification and treatment options for polycystic liver diseases. World J Gastroenterol. 2013; 19: 5775‑5786. | Crossref
- Zhang ZY, Wang ZM, Huang Y. Polycystic liver disease: classification, diagnosis, treatment process, and clinical management. World J Hepatol. 2020; 12: 72‑83. | Crossref
ARTICLE INFORMATION