Still diagnosed too late and under-recognized? The first comprehensive report on primary hyperoxaluria in Poland
Key words: clinical course, epidemiology, genetics, primary hyperoxaluria



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Still diagnosed too late and under-recognized? The first comprehensive report on primary hyperoxaluria in Poland
Introduction: Primary hyperoxalurias (PHs) are rare disorders leading to overproduction and increased urinary excretion of oxalate. Three monogenic forms (PH1–PH3) were classified. PHs lead to urolithiasis and chronic kidney disease. There are only sparse data on patients with PH from Eastern European countries including Poland.
Objectives: The aim of the study was to evaluate the prevalence, genetic background, and clinical course of PH in the Polish population.
Patients and methods: This was a retrospective multicenter study including data of all identified and genetically confirmed Polish patients with PH.
Results: Between 1998 and 2019, 21 patients with PH were identified, including 13 patients with PH1 (62%), 2 with PH2 (9%), and 6 with PH3 (29%). In those with PH1, the most common mutation was c.508G>A in AGXT and in PH3, c.700+5G>T in HOGA1. Nine patients (69%) developed end‑stage renal disease at a median age of 13 years and 2 died. In 6 (46%) PH1 cases, the diagnosis was made only after patients had progressed to end‑stage renal disease and received isolated kidney transplantation, followed by graft failure. Combined liver‑kidney transplantation was performed in 6 patients with PH1. Two siblings with PH2 showed a milder course with slightly decreased renal function in one, at age of 11 years. Despite infantile onset of urolithiasis, all patients with PH3 at a median age of 10 years maintained normal renal function.
Conclusions: The prevalence of PH1 and PH2 in Poland seems to be much lower than in Western countries with PH3 constituting about 30% of all cases. The molecular findings and clinical course are typical, but the underdiagnosis is of concern.
What's new?
This is the first comprehensive report on primary hyperoxaluria (PH) from Poland as well as from the Central Eastern / Eastern European countries. To date, the prevalence, clinical course, genetic background, treatment, and outcomes of this rare inherited metabolic disease in this region were completely unknown and only single case reports were published. The results indicate that the prevalence of PH in Poland is much lower than in Western countries and the distribution of PH subtypes differs from data demonstrated in North American and European registries. Although in the last decades considerable efforts have been made to facilitate diagnosis and improve the outcome of Polish patients with PH, delay in definite diagnosis is still a matter of concern.
Introduction
Primary hyperoxalurias (PH) are a group of autosomal recessive disorders of glyoxylate metabolism leading to endogenous overproduction and increased urinary excretion of oxalate. To date, 3 monogenic forms of PH (PH1–PH3) have been classified.
Primary hyperoxaluria type 1 (PH1; MIM, 259900) is the most common form identified worldwide and is caused by biallelic pathogenic variants of the AGXT gene, encoding the liver specific peroxisomal enzyme, alanine: glyoxylate aminotransferase (AGT). The enzyme catalyzes transamination of L‑alanine and glyoxylate to pyruvate and glycine. Its disturbed activity leads to cytosolic accumulation of glyoxylate which is further oxidized to oxalate and glycolate by cytosolic lactate dehydrogenase (LDH).1
Primary hyperoxaluria type 2 (PH2; MIM, 260000) is caused by biallelic pathogenic variants of the GRHPR gene, encoding the cytosolic / mitochondrial enzyme glyoxylate reductase / hydroxypyruvate reductase (GRHPR), mainly expressed in the liver. GRHPR catalyzes the transformation of glyoxylate to glycolate and hydroxypyruvate to D‑glycerate. Similarly to PH1, the excess of oxalate is derived from glyoxylate and in addition, deficiency of GRHPR leads to conversion of hydroxypyruvate to L‑glycerate.2 Primary hyperoxaluria type 2 accounts for approximately 9% of the PH cases identified in Europe, but is more prevalent in Asian populations.3,4
Primary hyperoxaluria type 3 (PH3; MIM, 613616) is associated with loss of the HOGA1 gene function. It encodes a mitochondrial enzyme, the 4‑hydroxy‑2‑oxo‑glutarate aldolase type 1 (HOGA), which is mainly expressed in the liver.5,6 HOGA plays a crucial role in hydroxyproline metabolism and catalyzes the cleavage of 4‑hydroxy‑2‑oxoglutarate (HOG) into glyoxylate and pyruvate. Therefore, its deficiency leads to mitochondrial accumulation of HOG which is shifted into the cytosol, where it is further transformed to glyoxylate by nonspecific cytosolic aldolase and finally to oxalate by cytosolic LDH.7 It has been proposed that HOG excess may also increase oxalate production by inhibition of GRHPR.6 Although PH3 accounts for approximately 6% to 10% of clinically presenting PH cases,3,8 carrier frequency data inferred from allelic frequencies of the most common pathogenic variants in the genome Aggregation Database (gnomAD) suggests that it may be the most common PH type in some populations, including Ashkenazi Jews and Caucasians.9 However, a markedly reduced penetrance of biallelic HOGA1 mutation carriers per se and the high rate of clinical remission with age result in exceptionally low numbers of identified adults with symptomatic PH3.5,9-12
Although the clinical spectrum of PH is variable, its main symptoms are related to recurrent calcium oxalate (CaOx) urolithiasis (UL), nephrocalcinosis (NC), subsequent tubulointerstitial inflammation and type‑specific risk of progressive chronic kidney disease (CKD).13,14 The most severe course and complications are seen in PH1 patients, as the majority of them will develop end‑stage renal disease (ESRD) over time.15 Some PH1 patients will already present with ESRD during infancy. The treatment with pyridoxine may significantly reduce urinary oxalate excretion and slow CKD progression in a subgroup of PH1 patients with homozygous c.508G>A (p.Gly170Arg) or c.454T>A (p.Phe152Ile) mutations of the AGXT gene.15-17 The progressive deterioration of glomerular filtration rate leads to oxalosis, a systemic disorder caused by multiorgan CaOx deposition.13 The outcome in PH2 is intermediate, but less benign than was formerly thought, since up to 25% of patients will develop ESRD in adulthood.16 In comparison to PH1/2, PH3 has significantly better prognosis with a high but not complete rate of clinical remission with age and a relatively low risk of advanced CKD.8,10,11
The currently available, definitive treatment option for PH1 patients with CKD stages 4 and 5 is combined or sequential liver and kidney transplantation (CLKT).18,19 For PH2 patients with ESRD the optimal transplantation strategy is less clear and most patients received isolated kidney transplantation (iKTx) with variable outcome.3
New therapeutic options based on RNA interference targeting hepatic key enzymes for oxalate precursor formation, including glycolate oxidase or hepatic LDHA are evaluated and constitute potential for a first precision medicine approach to the treatment of PH1 and PH2 in the near future.20,21
There is little data on PH patients from Central Eastern / Eastern European countries and only single case reports on Polish PH1 patients have been published as yet.22,23 Therefore, we systematically evaluated the prevalence, clinical course, genetic background, treatment, and outcome of PH in Poland.
Patients and methods
The project was designed as a retrospective multicenter study, to evaluate data of all identified and genetically confirmed Polish PH patients. All centers of pediatric nephrology and adult medical centers which diagnosed and reported PH cases to the national PH registry were included. The chart review comprised available selected anthropometric, clinical, and laboratory parameters (urinalysis, serum creatinine, urinary oxalate, calcium, and citrate excretion) obtained throughout individual follow‑up period.
In pediatric patients, estimated glomerular filtration rate (eGFR) was calculated using the original or modified Schwartz equation when applicable24,25 and in adults using the Modification of Diet in Renal Disease formula.26 Available urinary oxalate and citrate concentrations were measured enzymatically and their daily excretions were expressed in mmol per 1.73 m2 of body surface area. Daily urinary calcium excretions were calculated in mg per kg of body weight.
All molecular genetic analyses were performed at the Institute of Human Genetics, University of Cologne, Germany by targeted Sanger sequencing of the 3 disease associated genes AGXT, GRHPR, and HOGA1. Primer sequences and PCR conditions are available on request.
Statistical analysis
Statistical analysis was performed using the STATISTICA 13.1 software (StatSoft PL). Data were presented as median (range) as appropriate for continuous variables and as absolute numbers and / or percentages for categorical variables.
The study was approved by the Research Ethics Committee of Medical University of Lublin (no. KE‑0254/118/2016). An individual written consent was obtained from all adult patients or legal representatives of pediatric patients before molecular testing.
Results
The study included 21 individuals with PH from 15 families. They were diagnosed over a 21‑year period between 1998 and 2019. There were 13 PH1 (62%), 2 (9%) PH2, and 6 (29%) PH3 patients. All of them were Caucasians, and to the best of our knowledge, their parents were nonconsanguineous, though the degree of homozygosity calculated from available whole exome datasets of the 4 affected siblings from family 4 (F4; range 0.8% to 1.3%) would be close to the ratio of 1 to 64 expected for the offspring of second cousins. Their clinical characteristics are summarized in Table 1 and shown in detail in Tables 2 and 3.
PH1 (n = 13) | PH2 (n = 2) | PH3 (n = 6) | |
Data are presented as number (percentage) of patients unless otherwise indicated.
Abbreviations: CLKT, combined liver‑kidney transplantation; eGFR, estimated glomerular filtration rate; ESRD, end‑stage renal disease; FU, follow‑up; iKTx, isolated kidney transplantation; NC, nephrocalcinosis; PH1, primary hyperoxaluria type 1; PH2, primary hyperoxaluria type 2; PH3, primary hyperoxaluria type 3; UL, urolithiasis | |||
Age at the last FU, y, median (range) | 22 (3.5–72) | 12 (11–13) | 10 (7–13) |
Duration of FU, y, median (range) | 14 (1–44) | 10.5 (10–11) | 9 (7–12) |
Age at first symptoms, y, median (range) | 5 (1–28) | 1.5 (1–2) | 0.4 (0.3–0.7) |
First clinical symptoms | |||
UL | 4 (31) | 1 (50) | 6 (100) |
NC | 4 (31) | 1 (50) | 0 |
NC+UL | 5 (38) | 0 | – |
Hematuria | 3 (23) | 1 (50) | 5 (83) |
Sterile leukocyturia | 5 (38) | 1 (50) | 4 (67) |
Urinary tract infections | 4 (31) | 0 | – |
Decreased eGFR | 8 (62) | 1 (50) | 0 |
Age of clinical diagnosis / suspicion, y, median (range) | 13 (3.5–65) | 11.2 (10.5–12) | 3.2 (0.5–7.4) |
Time between first symptoms and clinical diagnosis, y, median (range) | 7 (0.2–37) | 9.8 (9.5–10) | 2.5 (0.1–7.1) |
Circumstances leading to diagnosis | |||
Metabolic evaluation of UL / NC | 5 (39) | 2 (100) | 6 (100) |
Evaluation of failed iKTx | 6 (46) | – | – |
Family screening of asymptomatic subjects | 2 (15) | – | – |
Selected urinary parameters at diagnosis | 7 (54) | 2 (100) | 6 (100) |
24h urinary oxalate excretion, mmol/1.73 m2, median (range) | 1.74 (1.05–3.72) | 1.6 (1.18–2.02) | 0.98 (0.73–1.6) |
24h urinary calcium excretion, mg/kg, median (range) | 0.87 (0.66–3.13) | 1.78 (0.95–2.62) | 2.46 (1–4.63) |
24h urinary citrate excretion, mmol/1.73 m2, median (range) | 0.79 (0.46–3.46) | 1.96 (1.47–2.45) | 2.23 (1.79–3.31) |
Age at molecular diagnosis, y, median (range) | 14 (3.5–65) | 12 (11–13) | 5 (0.6–8) |
Time between clinical and genetic diagnosis, y, median (range) | 0.5 (0.2–12) | 0.8 (0.5–1) | 0.4 (0.2–5.5) |
ESRD during FU | 9 (69) | 0 | 0 |
Age at ESRD, y, median (range) | 13 (2.5–63) | – | – |
Symptoms of systemic oxalosis | 7 (54) | 0 | 0 |
Organ transplantation | |||
Any | 8 (62) | 0 | 0 |
iKTx (n = 8) | 6 (75) | – | – |
CLKT after failed iKTx (n = 6) | 4 (67) | – | – |
Primary CLKT (n = 8) | 2 (25) | – | – |
Mortality | |||
All‑cause | 2 (15) | 0 | 0 |
Age at death, y, median (range) | 23 (22–24) | – | – |
Patient ID | Gender | Total FU time, y | Age at first symptoms, y | First symptoms | FU till diagnosis | Age of clinical diagnosis, y/date | Circumstances leading to diagnosis | FU after diagnosis | Outcome at the last FU | Genotype AGXT |
a Due to adoption, siblings F4.1/F4.2 and F4.3/F4.4 lived in separated families
Abbreviations: ARF, acute renal failure; CAPD, continuous ambulatory peritoneal dialysis; CKD, chronic kidney disease; ↓, decreased; ESWL, extracorporeal shock wave lithotripsy; HD, hemodialysis; KT, kidney transplant; LT, liver transplant; UTI, urinary tract infection; URS, ureterolithotripsy; others, see Table 1 | ||||||||||
F1 | F | 26 | 9 | NC, UTI, ↓eGFR | Progressive CKD, ESRD at 10 y, HD, iKTx at 13 y | 15/1998 | Failed iKTx (graft oxalosis) | HD, CLKT at 19 y, bone oxalosis | Chronic LT and KT failure | c.33delC(p.Lys12Argfs*34); c.33delC(p.Lys12Argfs*34) |
F2 | M | 14 | 8 | UL, UTI | Recurrent UL, multiple urological procedures (ESWL), progressive CKD, ESRD at 13 y, HD, iKTx at 17 y | 17/2000 | Failed iKTx (graft oxalosis) | HD, CLKT at 20 y, 3rd iKTx at 22 y, bone oxalosis | Death at 22 y (uremia complications) | c.508G>A(p.Gly170Arg); c.508G>A(p.Gly170Arg) |
F3 | M | 22 | 2 | NC, ↓eGFR | Progressive CKD, ESRD at 3 y, HD, CADO, iKT at 6 y | 7/2002 | Failed iKTx (graft oxalosis) | HD, CLKT at 8 y, bone oxalosis | Good LT function, chronic KT failure | c.533G>A(p.Cys178Tyr); c.533G>A(p.Cys178Tyr) |
F4.1a | F | 14 | 12 | UL / NC, sterile leukocyturia, ↓eGFR | Progressive CKD, progressive NC | 13/2005 | Metabolic evaluation | Alkaline citrate, fluids, pyridoxine resistant, recurrent UL, multiple urological procedures (ESWL, URS), progressive NC, progressive CKD, ESRD at 15 y, HD, CLKT at 16 y | Good LT and KT function | Homozygous large deletion exons 6 to 8 (p.Ile200Alafs*29) |
F4.2a | F | 11 | 24 | After diagnosis | Asymptomatic | 13/2005 | Family screening | Alkaline citrate, fluids, pyridoxine resistant, asymptomatic till 24 y | Urosepsis and ARF during pregnancy, HD, death at 24 y (oxalate cardiomyopathy) | Homozygous large deletion exons 6 to 8 (p.Ile200Alafs*29) |
F4.3a | M | 18 | 1 | NC, sterile leukocyturia, ↓eGFR | Progressive CKD, ESRD at 6 y, HD, iKTx at 8 y | 8/2008 | Failed iKTx (graft oxalosis) | HD, CLKT at 9 y | Good LT function, chronic KT failure | Homozygous large deletion exons 6 to 8 (p.Ile200Alafs*29) |
F4.4a | F | 11 | 21 | After diagnosis | Asymptomatic | 11/2009 | Family screening | Alkaline citrate, fluids, pyridoxine resistant, asymptomatic till 21 y – renal colic, ¯eGFR | CKD (eGFR 80ml/min/1.73 m2), solitary renal calcifications | Homozygous large deletion exons 6 to 8 (p.Ile200Alafs*29) |
F5 | F | 10 | 4 | UL / NC, UTI, ↓eGFR | Urological procedures (ESWL) | 4.3/2009 | Metabolic evaluation | Alkaline citrate, fluids, pyridoxine resistant, urological procedures (URS, ESWL),progressive CKD, ESRD at 10 y, CAPD, CLKT at 10.4 y | Good LT and KT function | c.508G>A(p.Gly170Arg); c.33dupC(p.Lys12Glnfs*156) |
F6 | F | 44 | 28 | UL, UTI | Multiple urological procedures (ESWL, unilateral nephrectomy), progressive CKD, ESRD at 63 y, HD, iKTx at 65 y | 65/2010 | Failed iKTx (graft oxalosis) | HD | On HD | c.508G>A(p.Gly170Arg); c.508G>A(p.Gly170Arg) |
F7.1 | F | 14 | 5 | UL / NC, sterile leukocyturia, hematuria, ↓eGFR | Conservative treatment of UL, slow progressive CKD | 17/2016 | Metabolic evaluation | Alkaline citrate, fluids, pyridoxine resistant, slow progressive CKD | CKD (eGFR 79 ml/min/1.73 m2) | c.603C>A(p.Asp201Glu); c.942+1G>T(p.?) |
F7.2 | F | 10 | 1 | UL / NC, sterile leukocyturia, hematuria, ↓eGFR | Conservative treatment of UL, progressive CKD | 13/2016 | Metabolic evaluation | Alkaline citrate, fluids, pyridoxine resistant, progressive CKD | CKD (eGFR 62 ml/min/1.73 m2) | c.603C>A(p.Asp201Glu); c.942+1G>T(p.?) |
F8 | M | 21 | 4 | UL / NC, sterile leukocyturia, ↓eGFR | Multiple urological procedures (ESWL), progressive CKD, ESRD at 19 y, HD, iKT at 21 y | 25/2018 | Failed iKTx (graft oxalosis) | HD | On HD, CLKT candidate, bone oxalosis | c.409C>T(p.Gln137*); c.942+1G>T(p.?) |
F9 | M | 1 | 2.5 | UL | Conservative UL treatment | 3.5/2019 | Metabolic evaluation | Alkaline citrate, fluids, response to pyridoxine not established yet | Normal eGFR | c.121G>A(p.Gly41Arg); c.697C>T(p.Arg233Cys) |
Patient ID | Gender | Total FU time, y | Age at first symptoms, y | First
symptoms | FU till diagnosis | Age at clinical
diagnosis, y/date | Circumstances leading to diagnosis | FU after diagnosis | Outcome at the last FU | Genotype GRHPR |
a Both missense variants c.284G>A and c.376G>A are found in cis on the paternal allele and occur in trans with the maternal c.700+5G>T splice variant
| ||||||||||
PH2 | ||||||||||
F10.1 | F | 12 | 1 | NC, sterile leukocyturia, hematuria, ↓eGFR | Recurrent UL since 9 y, multiple, urological procedures (ESWL, RIRS), conservative treatment (diet, fluids) | 10.9/2018 | Metabolic evaluation | Conservative treatment (diet, fluids) | CKD (eGFR 85 ml/min/1.73 m2) | c.103delG(p.Asp35Thrfs*11); c.454dupA(p.Thr152Asnfs*39) |
F10.2 | F | 11 | 2 | UL | Incidental UL, conservative treatment (diet, fluids) | 12/2019 | Metabolic evaluation | Conservative treatment (diet, fluids) | Normal eGFR | c.103delG(p.Asp35Thrfs*11); c.454dupA(p.Thr152Asnfs*39) |
PH3 | ||||||||||
F11 | M | 12 | 0.4 | UL, UTI, hematuria | – | 0.5/2005 | Metabolic evaluation | Conservative treatment (alkaline citrate, diet, fluids), urological procedure (ESWL), no UL recurrence, intermittent HC | Normal eGFR, no UL recurrence | HOGA1 homozygous c.700+5G>T(p.?); c.700+5G>T(p.?) |
F12 | F | 10 | 0.3 | UL, sterile leukocyturia, hematuria | – | 0.6/2012 | Metabolic evaluation | Conservative treatment (alkaline citrate, diet, fluids), urological procedure (URS), no UL recurrence, intermittent HC | Normal eGFR, no UL recurrence | c.700+5G>T(p.?); c.289C>T(p.Arg97Cys) |
F13 | M | 7 | 0.3 | UL, sterile leukocyturia, hematuria | Conservative treatment (diet, fluids), urological procedures (URS) | 2/2012 | Metabolic evaluation | Conservative treatment (diet, fluids), no UL recurrence | Normal eGFR, no UL recurrence | c.700+5G>T(p.?); c.580G>A(p.Gly194Ser) |
F14.1 | F | 11 | 0.7 | UL, sterile leukocyturia, UTI, hematuria | Conservative treatment (diet, fluids), urological procedure (URS) | 7/2013 | Metabolic evaluation | Conservative treatment (alkaline citrate, diet, fluids), no UL recurrent, intermittent HC | Normal eGFR, no UL recurrence | c.700+5G>T(p.?); c.206T>G(p.Phe69Cys) |
F14.2 | F | 8 | 0.5 | UL, sterile leukocyturia, hematuria | Conservative treatment (diet, fluids), urological procedure (URS, PCNL) | 4.5/2013 | Metabolic evaluation | Conservative treatment (alkaline citrate, diet, fluids), urological procedures (ESWL), intermittent HC, recurrent UL | Normal eGFR, UL | c.700+5G>T(p.?); c.206T>G(p.Phe69Cys) |
F15 | F | 8 | 0.3 | UL | Diet, fluids, urological procedure (URS, open surgery) | 7.4/2017 | Metabolic evaluation | Diet, fluids, intermittent HC, recurrent UL, urological procedures (ESWL, PCNL) | Normal eGFR, UL | c.700+5G>T(p.?); c.284G>A(p.Arg95His and c.376G>A(p.Ala126Thr)a |
Thirteen patients (5 men, 8 women) from 9 families had PH1. Their median (range) age at last follow‑up was 22 (3.5–72) years. The median (range) age at onset of manifestations was 5 (1–28) years and the median (range) time of the follow‑up from onset was 14 (1–44) years. The distribution of initial symptoms was as follows: isolated UL (31%), isolated NC (31%), and combined UL / NC (38%). They were accompanied by microscopic hematuria (38%), sterile leukocyturia (38%), or urinary tract infections (31%). Decreased eGFR was initially present in 62% of PH1 patients. The median (range) age at clinical diagnosis of PH was 13 (3.5–65) years and it was made from 0.2 to 37 (median, 7) years after the first clinical symptoms.
In almost half (46%) of the PH1 patients (mainly in cases from far past), the diagnosis of PH was not made until they reached ESRD and received iKTx (F1, F2, F3, F4.3, F6, F8). In these 6 cases, only rapidly progressive kidney graft failure and massive CaOx deposition within the renal graft tissue led to the correct diagnosis (Figure 1). Moreover, 4 of them had severe bone pain due to bone oxalosis (F1, F2, F3, F8).
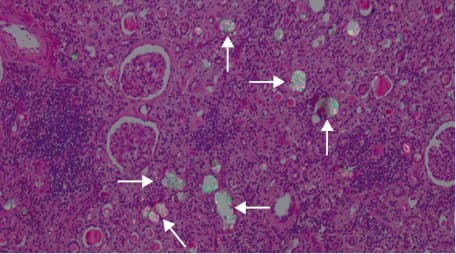
Only 7 PH1 patients (54%) were properly identified by metabolic screening for urinary oxalate. There were 5 (39%) patients with UL / NC and preserved own renal function (F4.1, F5, F7.1, F7.2, F9) and 2 (15%) clinically asymptomatic but biochemically affected siblings from PH1 F4 (F4.2, F4.4). The biochemical work‑up studies in these cases showed hyperoxaluria (HOx) which exceeded 1 mmol/1.73 m2/24 h (median [range], 1.74 [1.05–3.72] mmol/1.73 m2/24 h). In most of them, it was accompanied by decreased urinary calcium and citrate excretion (median [range] 0.87 [0.66–3.13] mg/kg/24 h and 0.79 [0.46–3.46] mmol/1.73 m2/24 h, respectively).
Genetic testing in the PH1 group revealed 8 patients with compound‑heterozygous and 5 with homozygous pathogenic AGXT variants. The most frequent mutation was c.508G>A (p.Gly170Arg). It affected 3 unrelated patients, of whom 2 were homozygous (F2, F6) and 1 (F5) compound‑heterozygous: c.508G>A (p.Gly170Arg); c.33dupC (p.Lys12Glnfs*156). Three patients from 2 families were heterozygous for the splice mutation c.942+1G>T (F7.1, F7.2, F8). In 2 families, the novel mutation c.533G>A (p.Cys178Tyr) (F3) and a novel large out‑of‑frame deletion of exons 6 to 8 (p.Ile200Alafs*29; F4.1, F4.2, F4.3, F4.4) were identified. In patient F1, c.33delC (p.Lys12Argfs*34) was identified in a homozygous state and patient F9 showed the 2 mutations c.121G>A (p.Gly41Arg) and c.697C>T (p.Arg233Cys) in a compound‑heterozygous state.
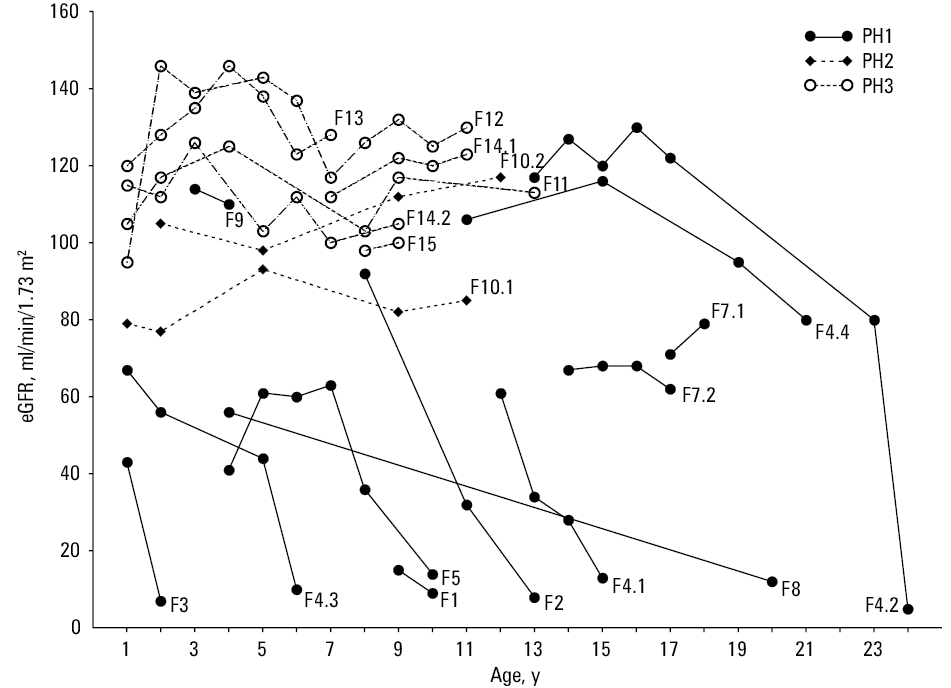
During follow‑up, patients with PH1 showed a variable course of CKD (Figure 2). Nine of them (69%) developed ESRD at a median (range) age of 13 (2.5–63) years. Two reached ESRD in early childhood (F3, F4.3), 5 during adolescence (F1, F2, F4.1, F5, F8), and 2 in adulthood (F4.2, F6). At the last follow‑up, 4 patients had preserved native kidney function, including 3 with CKD stage 2 (F4.4, F7.1, F7.2) and 1 with CKD stage 1 (F9). Treatment with pyridoxine was attempted only in 6 patients with preserved renal function (F4.1, F4.2, F4.4, F5, F7.1, F7.2) but it was ineffective and yielded no significant reduction in urinary oxalate excretion.
CLKT was performed in 6 PH1 patients. It was the primary procedure only in 2 patients (F4.1, F5) who were correctly diagnosed with PH1 before reaching ESRD. They received CLKT after several months of dialysis treatment. In the remaining 4 patients (F1, F2, F3, F4.3) with a delayed PH1 diagnosis, CLKT was performed 1 to 4 years after failed primary iKTx. At the last follow‑up, 5 patients treated with CLKT showed variable preserved liver and kidney graft function.
One patient (F2) died at the age of 22 years from uremia complications shortly after he received the third kidney graft, 2 years after CLKT. Patient F4.2 was clinically asymptomatic until the age of 24 years and presented with nonreversible acute renal failure caused by urosepsis at gestational week 27 of her third pregnancy. The patient died due to a late consequence of oxalate cardiomyopathy. Thus, the mortality rate in the PH1 group was 15%.
So far, only 2 patients (female siblings, F10.1, F10.2) with PH2 were identified. Their age at the last follow‑up was 11 and 13 years, respectively. Patient F10.1 presented with NC at 12 months, followed by recurrent UL, multiple urological interventions and CKD stage 2, whereas her sister showed incidental UL with a normal eGFR (Figure 2). Clinical suspicion of PH in these siblings was made only at the age of 10.9 and 12 years, when metabolic screening revealed HOx (2.02 and 1.18 mmol/1.73 m2/24 h, respectively). It was accompanied by increased urinary L‑glycerate (semiquantitative measurement) and normal calcium and citrate excretion. Genetic testing revealed 2 common pathogenic GRHPR variants: c.103delG (p.Asp35Thrfs*11) and c.454dupA (p.Thr152Asnfs*39) in compound‑heterozygous state.
Six patients (2 men, 4 women) from 5 families had PH3. Among those patients we could identify 3 novel causative HOGA1 variants: c.206T>G (p.Phe69Cys), c.580G>A (p.Gly194Ser), and c.284G>A (p.Arg95His) plus c.376G>A (p.Ala126Thr) in cis on the paternal allele. For the latter variants, it remains currently unclear which variant is deleterious or if potentially both variants result in loss of HOGA1 function. All novel variants occurred in trans with the common splice donor variant c.700+5G>T. Their median (range) age at the last follow‑up was 10 (7–13) years. The median (range) age at onset of symptoms was 0.4 (0.3–0.7) years and the median (range) time of the follow‑up from onset was 9 (7–12) years. Initially, all patients presented with UL, which was accompanied by microscopic hematuria (83%) or / and sterile leukocyturia (67%). The median (range) age at clinical suspicion of PH was 3.2 (0.73–7.4) years and the median (range) time of diagnostic delay was 2.5 (0.1–7.1) years. Comparing with patients with PH1/2, urinary oxalate excretion in PH3 cases was considerably lower (median [range] 0.98 [0.73–1.6] mmol/1.73 m2/24h). It was mostly accompanied by normal calcium and citrate excretion; however, on follow‑up, all but one (F13) patient showed intermittent moderate hypercalciuria (4–4.63 mg/kg/24 h). In all patients, genetic testing revealed the common c.700+5G>T splice site mutation: 1 patient was homozygous (F11) and the remaining 4 carried the splice mutation in a compound‑heterozygous state. After diagnosis, most patients were treated conservatively with alkaline citrate, a low‑oxalate diet, and high fluid intake. On the follow‑up, all patients had normal renal function (Figure 2). Only 2 patients (F14.2; F15) showed recurrent UL and required urological procedures.
Discussion
Until the late 1990s, patients with PH have not been identified in Poland, mainly due to limited access to relevant diagnostic tools. However, for more than 2 decades, considerable efforts have been made to facilitate diagnosis and improve outcomes. These included education of professionals, access to urinary oxalate measurement, and genetic evaluation through national and pan‑European initiatives like the European Hyperoxaluria Consortium (Oxaleurope). Accordingly, the vast majority of children and adolescents with UL / NC evaluated in all Polish centers of pediatric nephrology could be screened for HOx and all suspected pediatric and adult individuals were genetically tested. However, over the period 1998 to 2019, only 21 patients with PH, including 13 with PH1, were identified. This places a lower bound on the prevalence of PH1 of about 3 cases per 10 million, that is, approximately 3- to 10‑fold lower than reported in Western European countries and calculations based on carrier frequencies found in the gnomAD.9,27,28 Obviously, this discrepancy may be at least partly influenced by an ascertainment bias. We cannot exclude that some individuals may be still undiagnosed or even already dead without a proper diagnosis. On the other hand, we report a substantial cohort of PH3 patients, which may indicate that our efforts to improve awareness for PH in Poland are effective. Therefore, one could doubt that the more severe PH1 phenotype is generally overlooked, while the milder PH3 type is diagnosed and reported.
In theory, the carrier frequency of deleterious AGXT variants in the ethnic Polish population could be lower than in Western Europe but this remains to be conclusively measured. This might also account for other countries from Eastern Europe, separated behind the Iron Curtain for several decades. The paucity of published data on PH1 cases from this region may indirectly support this hypothesis. Other reasons contributing to data showing lower prevalence of PH1 in Poland may be related to the rarity of consanguineous marriages and historically little emigration from regions with a higher burden of the disease, like North Africa or Pakistan.4,29,30
To date, about 200 AGXT gene mutations have been described.8 In this study we provide for the first time genotypes of the Polish population. The common pathogenic variant in our cohort was c.508G>A (p.Gly170Arg), affecting 3 patients, of whom 2 were homozygous and 1 compound‑heterozygous (c.508G>A;c.33dupC). It is in agreement with other studies, which showed that it is the most frequent mutation among Caucasians, found in up to 30% of mutant alleles. In contrast, the duplication c.33dupC shows no ethnic association, accounting for 12% to 13% of PH1 cases from different populations.8,31-33 The second common mutation was c.942+1G>T affecting 3 patients from 2 families. Notably, 2 novel AGXT mutations, the missense variant c.533G>A (p.Cys178Tyr) and a complete (out‑of‑frame) deletion of exons 6 to 8 were found in 2 (22%) of 9 families.
PH1 is characterized by an extreme clinical heterogeneity with respect to age of onset and course.34 As in other observations, the median age of PH1 onset in the Polish cohort was 5 years.35 The majority of patients showed a typical course and most of them developed ESRD during puberty. Others had either late‑onset PH1 with first symptoms in adulthood or infantile / early childhood form with NC and rapid progression to ESRD within the first years of life. Several large studies showed that the homozygous c.508G>A mutation may be associated with a milder renal disease.8,15-17 It was explained by residual catalytic activity of mitochondrial mistargeted AGT which can be partially rescued by treatment with pyridoxine, the cofactor of AGT.17 This favorable course of patient F6 who presented very late with ESRD in the seventh decade of life in the absence of any specific treatment for PH may reflect these findings. In contrast, patient from F2 with the same genotype, also untreated with pyridoxine due to late diagnosis, developed ESRD at the age of 13 years and died 9 years later as a consequence of uremic complications. This characteristic for PH1 genotype‑phenotype discordance was even more impressive among 4 siblings (F4.1–4) sharing the same, large homozygous deletion (E × 6_8 del). Whereas patient F4.3 developed infantile oxalosis and F4.1 typical adolescent form of the disease, patients F4.2 and F4.4 remained asymptomatic till adulthood. This phenomenon of diverse intrafamilial disease expression in PH1 was already reported but the underlying factors are still unclear.36-39
The early diagnosis of PH1 is crucial for adequate therapeutic decisions and patients’ outcome. However, due to the rarity of this disease, lack of awareness or unusual presentation, it is often missed or diagnosis is delayed.40 In our group, the median time interval between initial symptoms and clinical diagnosis was 7 years. It seems to be significantly longer in comparison with the results of a German survey study from 2004 where two‑third of patients were diagnosed within 1 year.41 A dramatic example of diagnostic delay is the establishment of PH1 diagnosis only after recurrence of the disease in the isolated renal graft.22,42-46 In these cases, a graft failure occurs usually rapidly and biopsy reveals CaOx crystalline nephropathy as a consequence of post‑transplant mobilization of accumulated oxalate from body stores. Unfortunately, this scenario was observed in 6 Polish PH1 patients (46%) treated before the era of genetic testing, which is much more frequent than in other observational studies.16,45-47 Lack of proper diagnosis before ESRD had different reasons, which were mentioned before. It is noteworthy that the first patient with PH1 and preserved renal function was diagnosed in Poland only in 2005. Since that time 6 new cases received a more timely diagnosis, 4 of them during metabolic evaluation of UL / NC and the remaining 2 by family screening. They showed urinary oxalate excretion greater than 1 mmol/1.73 m2/24 h, all but one very low calcium excretion of less than 1 mg/kg/24 h, and half of them hypocitraturia. Unfortunately, due to unfavorable genotype, they were unresponsive to pyridoxine. Therefore, according to recommendations, they were / are treated symptomatically with high fluid intake, alkaline citrates, and low oxalate diet.18 Unfortunately, 2 of them reached ESRD and underwent CLKT (F4.1; F5) and one (F4.2), asymptomatic till 24 years, developed nonreversible acute renal failure caused by urosepsis in pregnancy and passed away due to a consequence of oxalate cardiomyopathy, awaiting combined heart‑liver‑kidney transplantation. The remaining 3 (F4.4; F7.1; F7.2), currently at the age between 16 and 22 years, are at CKD stage 2.
To date, 6 Polish PH1 patients underwent CLKT and their outcome has been already described.48 Timing of liver transplantation in PH1 is a matter of debate and preemptive procedure could potentially avoid complications of systemic oxalosis, but this advantage has to be carefully balanced against substantial perioperative risks.18 This strategy has not been applied to Polish patients. CLKT was the primary procedure in 2 patients who were correctly diagnosed with PH1 before reaching ESRD, whereas in the remaining 4 “historical” patients with delayed PH1 diagnosis, CLKT was performed 1 to 4 years after failed primary iKTx. Fortunately, due to specific treatment, including intensive hemodialysis, rapid renal graft loss was observed only in 1 of them (F2).48 As expected, most of these patients presented symptoms of systemic oxalosis, mainly bone pain. All but one patient preserved satisfactory kidney and liver function on the last follow‑up. Patient F2 lost his renal graft function and died with preserved liver function due to uremia complications after an unsuccessful third KTx at the age of 22 years.
Our data suggest that PH2 is extremely rare in Poland. Just recently, the first PH2 family with 2 affected siblings was identified. However, this type of PH may be associated with a greater problem of underdiagnosis in comparison with PH1 due to less severe phenotype.13 In Polish patients, 2 mutations in the GRHPR gene were found in a compound heterozygous state (c.103delG;c.454dupA). Despite the identical genotypes, both siblings showed a markedly different clinical course. The index case F10.1 presented with NC at 12 months, which was followed by recurrent UL, multiple urological interventions, and CKD stage 2, while her sister (F10.2) showed incidental UL with normal eGFR. The more severe phenotype in the former was associated with a significantly higher degree of hyperoxaluria. Interestingly, intrafamilial discordance in PH2 patients harboring the c.103delG mutation of the GRHPR gene was already reported.8 Thus, similarly to PH1, disease modifiers in PH2 should be also expected. In contrast to PH1, limited genotype‑phenotype correlations for PH2 could be established.3,8
Between 2012 and 2018, 6 patients with PH3 from 5 families were identified and they account for 29% of all cases in the whole PH cohort. This higher ratio compared with international data8 is the result of the abovementioned, remarkably low number of reported PH1 patients in Poland. In comparison with our PH1 group, the accurate clinical and molecular diagnosis in PH3 patients was established significantly faster, which might reflect an improvement in the clinical management of PH in Poland. However, considering the relatively short time from the first description of PH3, the self‑limiting course with age observed in many patients and unique appearance of ESRD, this type of PH is particularly prone to underestimation.5,12
In all Polish PH3 patients, at least one c.700+5G>T HOGA1 allele was found. This is the most common mutation identified in individuals of European descent with an allelic frequency up to 67% among causative variants.12,49
The clinical course in Polish PH3 patients was generally similar to that in other studies.8,12,49 It was characterized by very early onset with development of mostly bilateral UL in early infancy, usually accompanied by hematuria and sterile leukocyturia. The majority of patients required urological interventions within the first years of life. In contrast to PH1/2 patients, NC was occasionally reported,12,49,50 but was not observed in our PH3 group. During follow‑up, only 2 Polish PH3 patients showed recurrent UL and all of them had preserved normal renal function. Potential factors contributing to a favorable interim outcome in our cohort were a lower oxaluria observed in PH3 cases compared to the PH1 / PH2‑group, normocitraturia, early diagnosis, and timely introduction of conservative treatment, as well as avoidance of stone removal procedures. As reported previously,7,12,49 most Polish PH3 patients showed intermittent, moderate HC. This is in contrast to PH1 where low calcium excretion is a common finding, caused by massive CaOx precipitation.50
Conclusions
This is the first comprehensive study on PH from an Central Eastern / Eastern European country. The prevalence of PH1 and PH2 in Poland seems to be much lower than in many Western countries, while a substantial fraction of PH3 cases could be identified. The observed distribution of PH subtypes among Polish patients differs from the data reported by international registries. However, we cannot exclude at the moment that those differences may be largely influenced by the small sample size of the Polish cohort.
The molecular findings and clinical course of Polish PH patients are typical but a definite diagnosis of PH1 is still delayed, so further efforts among urologists and nephrologists are needed to increase the care and attention for the cohort with this rare disorders. In particular, urinary oxalate measurement should be a part of metabolic evaluation of every pediatric UL or NC case, as well as it should be performed in adults with recurrent UL or existing, unexplained NC.
- Danpure CJ. Molecular aetiology of primary hyperoxaluria type 1. Nephron Exp Nephrol. 2004; 98: e39‑44. | Crossref
- Cregeen DP, Williams EL, Hulton S, Rumsby G. Molecular analysis of the glyoxylate reductase (GRHPR) gene and description of mutations underlying primary hyperoxaluria type 2. Hum Mutat. 2003; 22: 497‑506. | Crossref
- Garrelfs SF, Rumsby G, Peters‑Sengers H, et al. Patients with primary hyperoxaluria type 2 have significant morbidity and require careful follow‑up. Kidney Int. 2019; 96: 1389‑1399. | Crossref
- Talati JJ, Hulton SA, Garrelfs SF, et al. Primary hyperoxaluria in populations of Pakistan origin: results from a literature review and two major registries. Urolithiasis. 2018; 46: 187‑195. | Crossref
- Belostotsky R, Seboun E, Idelson GH, et al. Mutations in DHDPSL are responsible for primary hyperoxaluria type III. Am J Hum Genet. 2010; 87: 392‑399. | Crossref
ARTICLE INFORMATION