Endoplasmic reticulum stress and proteasome inhibitors in multiple myeloma: room for improvement
Key words: endoplasmic reticulum stress, myeloma, proteasome inhibitors, resistance, unfolded protein response



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Endoplasmic reticulum stress and proteasome inhibitors in multiple myeloma: room for improvement
In the last 2 decades, we witnessed unprecedented progress in multiple myeloma research. The median survival times doubled, and with the introduction of subsequent new therapeutics, we expect even better results in the nearest future. However, the disease still remains incurable. It is attributed to recurring nature of multiple myeloma with reappearance of subclones resistant to previously used therapies. More than 15 years after the approval of the first‑in‑class proteasome inhibitor, bortezomib, the mechanisms responsible for resistance to this class of drugs are still not fully elucidated. One of the most promising explanations involves modulation of endoplasmic reticulum stress caused by accumulation of misfolded proteins. Due to excessive monoclonal protein production, multiple myeloma cells are particularly susceptible to proteotoxicity. Under normal circumstances, they counteract it with activation of an adaptive mechanism, that is, the unfolded protein response. This pathway, however, can also lead to cell apoptosis when unable to restore proteostasis. It is the expected effect of proteasome inhibition. Resistant cells develop mechanisms that decrease the endoplasmic reticulum stress. This review covers current efforts to understand the nature of this adaptation. It focuses on druggable targets that can potentially enhance proteasome inhibitors activity or resensitize resistant patients to this type of therapy.
Introduction
Multiple myeloma (MM) is one of the most common hematologic malignancies in Western countries, responsible for over 10 000 deaths per year in the United States.1 Despite tremendous improvement in patient prognosis achieved in the last 2 decades, the disease is still considered incurable in most cases.2-4 This situation is attributable mostly to the recurring nature of MM. The vast majority of patients with newly diagnosed MM (NDMM) respond well to first‑line therapy with modern drugs and achieve remission that can last a few years,5-7 but eventually, almost all of them experience clinical relapse. Treatment of refractory / relapsed (RR) disease is not as efficient as of the NDMM, and every subsequent line of therapy yields poorer results.8,9 To overcome this limitation, 2 approaches can be utilized: 1) using classes and types of drugs that were not administered in the previous lines of therapy or 2) adding agents that can augment activity of previously used medications and resensitize MM cells to them. Currently, the first approach is the only available and efficient way to treat patients with RR MM10,11 whereas the second one is still under development. Nevertheless, recently 2 drugs have been approved that—at least partially—utilize the concept of synergistic improvement. The first one, panobinostat, a deacetylase inhibitor that disrupts protein degradation by the aggresome pathway, has shown superiority in comparison with placebo when administered in combination with bortezomib and dexamethasone in the phase 3 PANORAMA (Panobinostat or Placebo With Bortezomib and Dexamethasone in Patients With Relapsed Multiple Myeloma) trial.12 Intriguingly, the drug does not exhibit single‑agent activity, which emphasizes the synergistic effect with proteasome inhibition. Similarly, the second drug, selinexor, which inhibits nuclear export through exportin‑1 (XPO1) has recently proved its efficacy in combination with bortezomib and dexamethasone.13 This is a particularly important finding in the context of the previously identified role of XPO1 overexpression in resistance to bortezomib.14 Thus, with the growing understanding of mechanisms responsible for resistance to the most common classes of anti‑MM drugs, it seems reasonable to expect that also the resensitizing / augmenting approaches will become effective and widely used. In the context of resistance to therapy, the most intensively studied class of anti‑MM drugs are proteasome inhibitors (PIs).15,16 Multiple myeloma cells are particularly susceptible to proteasome inhibition due to overproduction of monoclonal protein that requires high activity of the ubiquitin‑proteasome system (UPS).17 When the UPS is overwhelmed, another important adaptive reaction is involved in maintaining cell homeostasis, namely the unfolded protein response (UPR) triggered by endoplasmic reticulum (ER) stress.18,19 In this review, we summarize current knowledge on the role of ER stress modulation in myeloma cells resistance to PIs with special attention to druggable targets that can augment therapeutic efficacy of PIs.
Endoplasmic reticulum stress and unfolded protein response
Posttranslational modification (ie, disulfide bond formation, glycosylation) and folding of all secretory proteins occur in the ER.20 The process is thoroughly controlled by various chaperones and through maintaining appropriate redox conditions and calcium concentration in the ER lumen. Only proteins that are properly folded pass quality control and are allowed to leave the compartment to perform their physiological function. The misfolded proteins are retrotranslocated and polyubiquitinated to be directed for degradation in proteasome.21 26S proteasomes use multiple proteases to break peptide bonds and degrade unneeded proteins. The process of utilization of misfolded proteins by the UPS is called ER‑associated degradation (ERAD) and is essential for maintaining cell proteostasis.22 Endoplasmic reticulum stress starts when the capability of the ER to fold proteins is lower than the load of polypeptides that enter the compartment. The stress can be triggered by an increase in protein influx (pancreatic β cells during an insulin‑resistant state, malignant myeloma cells), a decrease in folding capacity (nutrient deprivation, hypoxia, calcium concentration aberrations, alterations of redox homeostasis), or interruption of ERAD (proteasome inhibition).23 Because the accumulation of misfolded proteins induces potentially fatal proteotoxicity, the eucaryotic cells developed a highly conservative adaptive response system to counteract it, that is, the UPR.18-20,24 It is triggered by ER stress and is regulated by 3 ER transmembrane proteins: inositol‑requiring enzyme 1 (IRE1), double‑stranded RNA‑activated protein kinase–like ER kinase (PERK), and activating transcription factor 6 (ATF6). These enzymes are inactive under physiological conditions, without a corresponding increase in ER stress.18 They remain inactive thanks to the binding of binding immunoglobulin protein (BiP; or glucose‑regulated protein 78 [Grp78]), a heat‑shock protein and a chaperone, to the transmembrane proteins. During ER stress caused by the accumulation of unfolded proteins whose hydrophobic fragments have higher affinity to bind BiP, the inhibiting protein dissociates from IRE1, PERK, and ATF6, leading to their activation and initiation of the UPR. As a result, activation of certain mechanisms leading to the induced translation of proteins that can restore ER homeostasis is initiated whereas translation of other nonessential proteins is stopped.24 IRE1 dimerizes to induce its endoribonuclease activity and splices 1 x‑box binding protein (XBP1) to produce spliced XBP1 (XBP1s), a transcription factor responsible for initiating processes that lead to increased protein folding and degradation of misfolded proteins in the ER. The endoribonuclease does not splice exclusively XBP1 but can also cleave other RNAs in a process called regulated IRE1‑dependent decay, attributing to lowering the burden of proteins processed by ER. Activation of PERK leads to phosphorylation of eukaryotic translation initiation factor 2 (eIF2a) and subsequent repression of global protein translation while simultaneously allowing for selective translation of activating transcription factor 4 (ATF4), a transcription factor for proteins necessary in restoring ER homeostasis (antioxidants, chaperones, elements of phagosome). ER stress triggers ATF6 dissociation from the ER membrane and its further translocation to the Golgi apparatus. It is subsequently cleaved to reveal their transcription factor domain, ATF6f, through which it regulates expression of other UPR genes. Despite being an adaptive mechanism to help cells survive under ER stress conditions, in the event of overwhelming ER stress, the same mechanism switches toward apoptosis, leading to cell death.25 It is also mediated by downstream effects of IRE1 and PERK activation. When ER stress becomes unsustainable, the C/EBP homologous protein (CHOP) transcription factor is activated by PERK, leading to inhibition of antiapoptotic proteins from the B‑cell lymphoma 2 (Bcl‑2) family. Also IRE1‑dependent decay activity ultimately results in decomposition of RNA encoding ER stress–relieving proteins. Since the balance between adaptation and apoptosis in response to ER stress is very subtle, it is reasonable to utilize this vulnerability in MM treatment.
Endoplasmic reticulum stress in myeloma cells and the impact of proteasome inhibitors
Due to the extensive monoclonal protein synthesis, myeloma cells are exposed to significant ER stress and are therefore highly dependent on the activity of the UPR pathway.26,27 At least a single study has shown that a marker of UPR activation, a grp94 (also known as heat shock protein 90 kDa beta member 1 [HSP90B1]) chaperone exhibit a significantly high expression in cells of patients with MM in comparison with those with monoclonal gammopathy of undetermined significance and the healthy population28 underlining the role of UPR in the survival of myeloma cells. Under undisturbed conditions, the UPR activity sufficiently adapts myeloma cells to intensive protein production.29 This situation changes when MM cells are exposed to PIs. Drugs from this class bind to different proteasome subunits (β1, β2, β5) to stop their chymotrypsin-, caspase-, or trypsin‑like catalytic activities and disrupt ERAD by blocking the UPS pathway.30 This process increases ER stress to the point when UPR is not able to restore proteostasis. The overwhelmed UPR stops supporting adaptive mechanisms and activates proapoptotic pathways instead.31,32 This can partially explain high efficacy of PIs in the treatment of MM. With 3 different drugs from this class currently approved (bortezomib, carfilzomib, ixazomib), the PIs are the backbone of a majority of modern anti‑MM therapies.33 There are some differences among the 3 drugs—carfilzomib and ixazomib preferentially block β5 subunit, whereas bortezomib blocks β5 and β1. Moreover, only carfilzomib inhibits the proteasome irreversibly,30 but they all show similarly high antimyeloma efficacy. However, even using PIs in combination with lenalidomide in patients with NDMM, as shown in the recent ENDURANCE trial, is not sufficient to generate response in all of the MM patients.34 Between 5% to 10% patients are primarily resistant to the therapy and majority of those treated with PIs experience relapse that is resistant to this type of drugs.35
Adaptations in proteasome inhibitor–refractory cells
Despite the fact that there are certain resistance mechanisms that appear unique for different members of PI class (eg, important role of drug efflux pump p‑glycoprotein expression in resistance against carfilzomib, not affecting bortezomib‑resistant cells),36 it is postulated that one of the major factors responsible for failure of all 3 approved PIs therapies is that resistant cells are less dependent on UPR, showing lower baseline ER stress level. It was described in cell lines as well as in patient samples. Leung‑Hagesteijn et al37 described PI‑resistant plasma cells population as IRE1/XBP1s negative, making an argument that this arm of UPR is indispensable for bortezomib toxicity. They showed that this adaptation decreases ER stress by plasma cell decommitment towards earlier progenitors that produce less monoclonal protein. This finding is in line with a known function of XBP1s in plasma cells maturation,38 previous observations on the impact of the level of immunoglobulin production on bortezomib activity,17 as well as with the correlation of the bortezomib sensitivity in mantle cell lymphoma with cell secretory load.39 Also, clinical observations prove that patients producing higher amounts of monoclonal protein respond better to PI‑based therapy,40 that the presence of extramedullary disease is associated with poorer prognosis,41 and that a low XBP1s expression is an adverse prognostic factor for patients undergoing bortezomib‑based therapy.42,43 Interestingly, the latter conclusion is not true for patients treated with thalidomide, underpinning that this mechanism is PI‑specific.44 In a recent study, Zang et al45 linked the XBP1s‑low phenotype with the expression of cell division cycle 37 (Cdc37), a cochaperone of heat shock protein 90 (Hsp90), another chaperone stabilizing many oncogenes,46 and confirmed its impact on bortezomib resistance. They also provided interesting explanation of discrepancies between high Cdc37 expression in NDMM samples in contrast to low expression in bortezomib‑resistant samples from patients with RR MM, attributing it to the clonal selection,47 a common phenomenon that includes also evolution of cytogenetic lesions.48 On the contrary, Borjan et al49 assessed amounts of secreted proteins, misfolded proteins in ER, and markers of UPR activation (BiP and PERK), and were unable to show significant differences between bortezomib‑sensitive MM cell lines and solid tumors cells. They confirmed, however, that bortezomib‑sensitive cells show higher activity of the IRE1/XBP1 pathway but considered it solely as a marker of plasma cell differentiation, not correlated with ER stress. Soriano et al36 investigated differences between PI‑sensitive and PI‑resistant cells and showed that the latter are characterized by lower UPR activity and proposed a model of metabolic adaptations that enhance protein‑folding capacity of ER by maintaining redox homeostasis, leading to decreased ER stress. They further described the changes by metabolomic analysis and showed enrichment in glutathione and NADP(H) renewing pathways, tricarboxylic acid cycle that altogether translated into increased reactive oxygen species buffering capacity leading to more efficient protein folding.50 Also, a change in lipid synthesis from lysolipids to sphingomyelins was described in this study, consistent with previous reports on the role of lipid composition of ER membrane in ER stress modulation.51,52
Targeting endoplasmic reticulum stress to enhance the activity of proteasome inhibitors
A number of ways to augment the drug activity in MM cells have been proposed in the recent years, based on the better understanding of ER stress and UPR activation roles in resistance to PIs. Currently explored approaches are discussed in this section and summarized in Table 1. The role of target molecules in the context of ER stress in the context of ER stress and UPR modulation are shown in Figure 1.
Molecular target | Target’s function | Drug | Effect | Clinical relevance | Stage of research | References |
Abbreviations: BiP, binding immunoglobulin protein; ER, endoplasmic reticulum; Ero1, FAD‑containing endoplasmic reticulum oxidoreductin 1; Grp78, glucose regulated protein 78; GSTP, glutathione S‑transferase P; IRE, inositol‑requiring enzyme; MAPK, mitogen‑activated protein kinase; MM, multiple myeloma; PDI, protein disulphide isomerase; PERK, double‑stranded RNA‑activated protein kinase–like ER kinase; PI, proteasome inhibitors; SGK, serum and glucocorticoid‑regulated kinase; SK2, sphingosine kinase 2; UPR, unfolded protein response | ||||||
GSTP | BiP glutathionylation, increasing the chaperone’s activity | TLK199 | Resensitization of bortezomib‑resistant cells | S‑glutathionylation overrepresented in patients with MM | Cell lines | Zhang et al53 |
Ero1 | Disulfide bond formation | EN‑460 | Activity against MM cell lines | Increased Ero1 expression correlates with poorer responses to bortezomib | Cell lines | Hayes et al56 |
PDIs | Disulfide bond formation | E64FC26 | Resensitization of PI‑resistant cells | PDIs over‑accumulated in bortezomib‑refractory patients | Cell lines, mouse models | Robinson et al59 |
PERK | UPR activation | GSK2606414 | Enhances bortezomib activity | PERK overexpressed in patients with MM | Cell lines | Bagratuni et al60 |
IRE1a | UPR activation | MKC‑3946, compound 18 | Enhances bortezomib activity | IRE1a correlated with the survival of patients with MM | Cell lines, mouse models, patient‑derived CD138+ cells | Mimura et al61 and Harnoss et al62 |
Grp78 | ER stress initiation, chaperone | PAT‑SM6 | Activity against MM cell lines | Clinical activity described in a case report | Phase 1 clinical trial | Rasche et al65 and Rasche et al66 |
SGK | Protection against ER stress‑induced apoptosis | GSK650394 | Resensitization of PI‑resistant cells | Increased SGK expression correlates with poorer responses to bortezomib | Cell lines | Hoang et al67 |
MAPK | Enhancing proteasome activity, multiple other functions | Kinase inhibitors | Enhances the activity of bortezomib and carfilzomib | Commonly mutated genes in patients with MM | Phase 1 and 2 clinical trials | Shirazi et al72 |
SK2 | Sphingosine phosphorylation | ABC294640, K145 | Enhances bortezomib activity | SK2 overexpressed in patients with MM | Phase 1b/2 clinical trial | Venkata et al78 and Wallington‑Beddoe et al79 |
HIV protease | Proteasome inhibition, increasing ER stress, multiple other functions | Nelfinavir | Resensitization of PI‑resistant cells | Response to treatment documented in clinical trials | Phase 2 clinical trial | Driessen et al82 and Driessen et al83 |
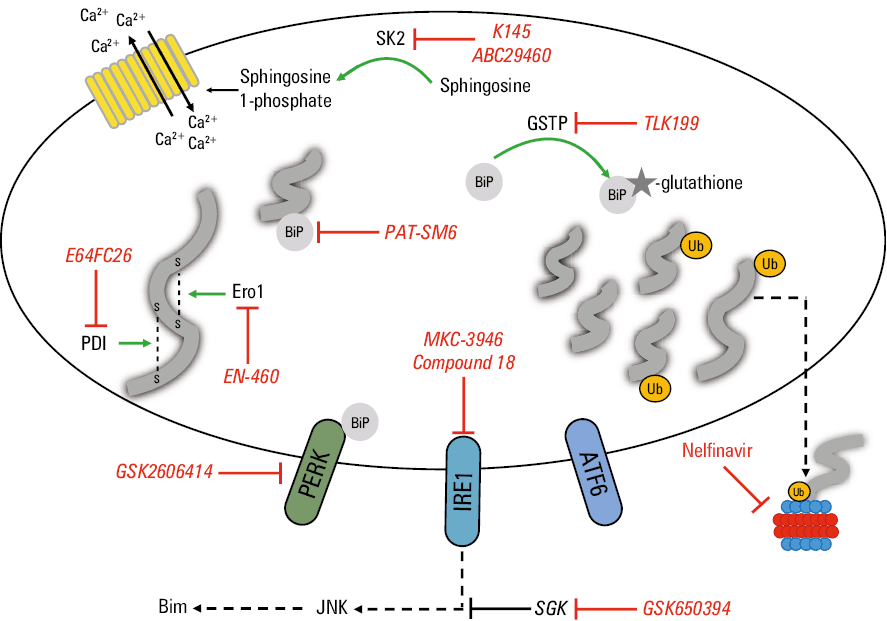
Abbreviations: ATF6, activating transcription factor 6; others, see Table 1
Zhang et al53 exploited the finding that bortezomib‑resistant cells show upregulation of the S‑glutathionylation level.54 They were able to identify BiP S‑glutathionylation by glutathione S‑transferase P (GSTP) as a way to increase the chaperone’s ability to fold proteins and therefore reduce ER stress and burden put on the UPS. Its clinical relevance was postulated by confirming higher BiP and S‑glutathionylation levels in bone marrow samples from patients with MM in comparison with healthy subjects. The study showed that GSTP inhibition by TLK199, a drug studied in a phase 1–2 clinical trials in myelodysplastic syndromes55 can resensitize MM cells to bortezomib.
Hayes et al56 identified FAD containing ER oxidoreductin 1 (Ero1) as another target to increase ER stress. The protein enables the formation of disulfide bonds in ER‑processed proteins, which leads to maintaining redox homeostasis. It is particularly important in MM cells because their ability to secrete proteins rich in disulfide‑bonds is among the highest in mammalian cells.57 The group proved that blocking Ero1 with specific inhibitor EN‑460 increases ER stress and exhibits promising anti‑MM activity in vitro. Although the study showed that increased Ero1 expression on MM cells is an adverse prognostic factor for patients treated with bortezomib, it remains to be determined if EN‑460 can enhance PI activity. Another approach exploiting similar vulnerability is the inhibition of proteins from the protein disulfide isomerase (PDI) group—oxidoreductase enzymes responsible for proper protein folding by creating disulfide bonds. Our proteomic studies confirmed increased accumulation of proteins from this family in a subgroup of bortezomib‑resistant patients.58 Robinson et al59 developed a new pan‑PDI inhibitor E64FC26 that showed promising activity in resensitizing bortezomib‑resistant cell lines. The effect was attributed to UPR activation and it was able to potentiate also the efficacy of other drugs from the PIs group. E64FC26 was also efficient in vivo, in mice xenotransplanted with human MM cells. Interestingly, combination of E64FC26 with bortezomib allowed for a greater increase in the median survival, in comparison with vehicle, than bortezomib alone.
Targeting the effector arms of UPR has also been developed as a way to potentiate the effect of proteasome inhibition. Bagratuni et al60 tested the activity of PERK inhibitor, GSK2606414 and proved its efficacy against human MM cell lines as well as synergistic effect when coadministered with bortezomib. The apoptotic effect was accompanied by an increase in expression of surviving, a gene associated with ER stress. Also inhibiting the IRE1/XBP1s axis of UPR showed encouraging activity against MM cell lines, mouse myeloma models and patient‑derived MM cells.61,62 Harnoss et al62 comprehensively described effects of IRE1a inhibition. They showed that it causes perturbations in the ERAD pathway and a decrease in the secretion of immunoglobulin light chains and cytokines, enabling anti‑MM effect. They were demonstrated that IRE1a blocking by compound 18 significantly reduces the viability of CD138+ cells from patients with MM while sparing CD138– population and not affecting homeostasis in tested mice. These findings advocate for further evaluation of IRE1a inhibitors in human clinical trials. However, it is important to acknowledge the complex involvement of IRE1 activation in MM pathogenesis. Goldsmith et al63 have recently proved in a large NDMM population (n = 768) that a high IRE1a expression is associated with significantly worse treatment outcomes: hazard ratio of 1.37 for progression and of 1.55 for death. Intriguingly, the study also showed that the expression of IRE1a in post‑treatment samples from refractory patients (disease progressed during treatment) is significantly lower than in paired pretreatment specimens. The authors, similarly to Zang et al,45 explain this counter‑intuitive finding by clonal selection. Thus, with this complexity in mind, efficacy of IRE1a inhibition in PI‑resistant setting remains to be determined.
Grp78, a chaperone with pleiotropic functions in ER stress response, from initiating the UPR to increasing ER folding capacities, is another promising target.64 Monoclonal IgM antibody against Grp78 was isolated and developed as PAT‑SM6. After showing in vitro activity, the product entered clinical phase of studies, showed favorable safety profile but failed to induce significant tumor reduction.65 Even though the drug’s further tests were stopped, a case report of successful treatment of extramedullary, RR disease using combination of PAT‑SM6 with bortezomib and lenalidomide66 may act as a proof of principle for enhancing PI activity in patients with RR MM by Grp78 inhibition.
Hoang et al67 described protective function of serum and glucocorticoid‑regulated kinase (SGK) in response to ER stress by disrupting the IRE1 / apoptosis signal‑regulating kinase 1 (ASK‑1) / c‑Jun N‑terminal kinase (JNK) pathway—the proapoptotic axis of UPR.68 The study showed that patients with higher SGK expression achieve worse response to therapy with bortezomib. Recently, Tsubaki et al69 showed that SGK inhibition by GSK650394 enhances bortezomib and ixazomib toxicity in primarily resistant MM cells.
Genes from the RAS / mitogen‑activated protein kinases (MAPK) pathway are most commonly mutated in MM,70 with NRAS mutations corresponding with decreased sensitivity to PIs.71 Shirazi et al72 linked activating KRAS, NRAS, and BRAF mutations with decreasing ER stress due to enhanced proteasome activity. The findings were supported by the confirmation of additive effect of using MAPK (selumetinib, trametinib) and RAF (TAK‑632) kinase inhibitors in combination with bortezomib or carfilzomib. It provides mechanistic rationale for the kinase inhibitors activity in PI‑resistant MM patients that has been recently described in case reports73,74 and is investigated in many phase 1 and 2 clinical trials.75
As previously mentioned, UPR can be activated by disturbances in membrane lipid saturation by interacting with the ER calcium level and therefore affecting ER folding capabilities.76 Interestingly, even in cells with mutant IRE1 and PERK, missing their BiP‑binding luminal domains, UPR can be initiated by the direct activation of sensors through lipid perturbations.77 In line with these findings, Venkata et al78 showed that sphingosine kinase 2 (SK2), one of the key enzymes in sphingolipids metabolism, is overexpressed in patients‑derived myeloma cells and that SK2 inhibitor, ABC294 640 has anti‑MM activity against cell lines and mouse MM models. A phase 1b/2 clinical trial of ABC294640 (ClinicalTrials.gov identifier, NCT02757326), aiming to accrue 13 RR MM patients was initiated in 2016 but terminated in 2019 due to expiration of funding. The results of this study have supposedly been submitted but are not publicly available yet. Wallington‑Beddoe et al79 were able to connect SK2 inhibitor activity with increasing ER stress and activating UPR. This finding was enforced by synergistic effect of combining bortezomib with another SK2 inhibitor, K145.
As for now, the most compelling data in favor of the idea of resensitizing patients to PIs were provided by the Swiss group led by Driessen who combined bortezomib with HIV protease inhibitor, nelfinavir. The drugs from this latter group, apart from being efficient in HIV infection treatment, also showed antineoplastic activity.80 Among their biological effects, proteasome inhibition and increasing ER stress were identified. These findings led the investigators to assess anti‑MM activity of HIV protease inhibitors. First, they were able to show that, in this class of drugs, nelfinavir has the highest in vitro efficacy and synergistic activity with bortezomib and carfilzomib.81 Then, a phase 1 trial of nelfinavir in combination with bortezomib identified 2 × 2500 mg dose as safe and potentially efficient: clinical activity was seen in 5 (3 partial responses, ie, >50% reduction of monoclonal protein, 2 minor responses, ie, >25% reduction of monoclonal protein) out of 6 bortezomib- and lenalidomide‑refractory patients treated in the extension cohort.82 The subsequent phase 2 trial also yielded promising results.83 The use of combination of nelfinavir, bortezomib and dexamethasone resulted in at least partial response in 65% (22/34) of the heavily‑pretreated patients with MM (median lines of previous therapies, 5; 100% bortezomib refractory, 100% lenalidomide exposed). The authors underlined that response rates in such a population of patients were similar or better than those achieved with recently approved novel drugs, whereas the estimated cost of the therapy does not exceed 15% of the medications currently used in this setting.
Conclusions
With the growing body of evidence supporting crucial interactions between ER stress, UPR, and PIs activities, translation of these results into clinically meaningful outcomes is eagerly awaited. Better understanding of myeloma cells biology in the context of proteasome inhibition leads to identification of many vulnerabilities that can potentially be targeted to improve current treatment results. At this moment, among the investigated particles able to target the ER homeostasis, only a minority entered the clinical trials phase. However, the example of nelfinavir provides proof of concept that adaptations present in resistant MM cells could be used in clinical practice. It should encourage the research community to further develop the resensitizing approach.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020; 70: 7‑30. | Crossref
- Fonseca R, Abouzaid S, Bonafede M, et al. Trends in overall survival and costs of multiple myeloma, 2000–2014. Leukemia. 2017; 31: 1915‑1921. | Crossref
- Thorsteinsdottir S, Dickman PW, Landgren O, et al. Dramatically improved survival in multiple myeloma patients in the recent decade: results from a Swedish population‑based study. Haematologica. 2018; 103: e412‑e415. | Crossref
- Bazarbachi AH, Al Hamed R, Malard F, et al. Relapsed refractory multiple myeloma: a comprehensive overview. Leukemia. 2019; 33: 2343‑2357. | Crossref
- Paquin AR, Kumar SK, Buadi FK, et al. Overall survival of transplant eligible patients with newly diagnosed multiple myeloma: comparative effectiveness analysis of modern induction regimens on outcome. Blood Cancer J. 2018; 8: 125. | Crossref
ARTICLE INFORMATION