Hajdu‑Cheney syndrome (HCS) is a very rare genetic disorder (less than 50 cases described to date) of an autosomal dominant inheritance, or developing as a result of a de novo mutation in the NOTCH2 gene encoding the notch2 protein. The mutation results in an enhanced notch2 signaling.1 Notch is a transmembrane protein involved in several physiological processes, contributing to the cell fate determination and critically influencing tissue development. Notch2 positively regulates receptor‑activated nuclear factor–κB ligand–induced osteoclastogenesis which results in bone deformations upon enhancement of this signaling pathway.1
The disease affects many body systems with predominant skeletal involvement. Acro‑osteolysis is most characteristic of this disease, but other health issues can occur including osteoporosis, skull abnormalities (eg, facial bones), dental problems, hearing loss, deep gravelly voice due to recurrent infections, cardiac defects, and kidney abnormalities (mainly multiple renal cysts). Most serious manifestations include: platybasia (abnormal flattening of the skull base) and basilar invagination. These complications can lead to severe headaches, hydrocephalus, breathing disorders, and even sudden death.2-5
A woman aged 21 years at presentation was diagnosed with HCS at the age of 6 years. The diagnosis was based on several imaging techniques that revealed a dolichocephalic skull and the typical facial features: widely spaced eyes, low‑set ears, and midface hypoplasia. Other abnormalities included: patent ductus arteriosus (closed shortly after birth), multiple renal cysts, hearing loss, and bone lesions. The latter comprised: wormian bones, platybasia, widened sella turcica, abnormally shaped (shortened and arched) all long bones, pathological fractures of metatarsal bones, acro‑osteolysis of phalanges, and delayed bone age (by 5 years). Vertebral lesions were also noticed: biconcave “fish” vertebrae and spondylolisthesis. The medulla oblongata and pons were modelled from the front by bone structures. Other symptoms included hydrocephalus and syringomyelia. The mentioned kidney disorder led to chronic and then end‑stage renal failure at the age of 10, and consequently, to a kidney transplant at the age of 12. The patient suffered from recurring respiratory infections and finally respiratory failure that required mechanical ventilation. She died at the age of 21 due to respiratory failure and cardiac arrest. Genetic testing (search for mutation) was not done because of a very typical clinical and imaging phenotype. All the deformities are presented in Figure 1A–1F.
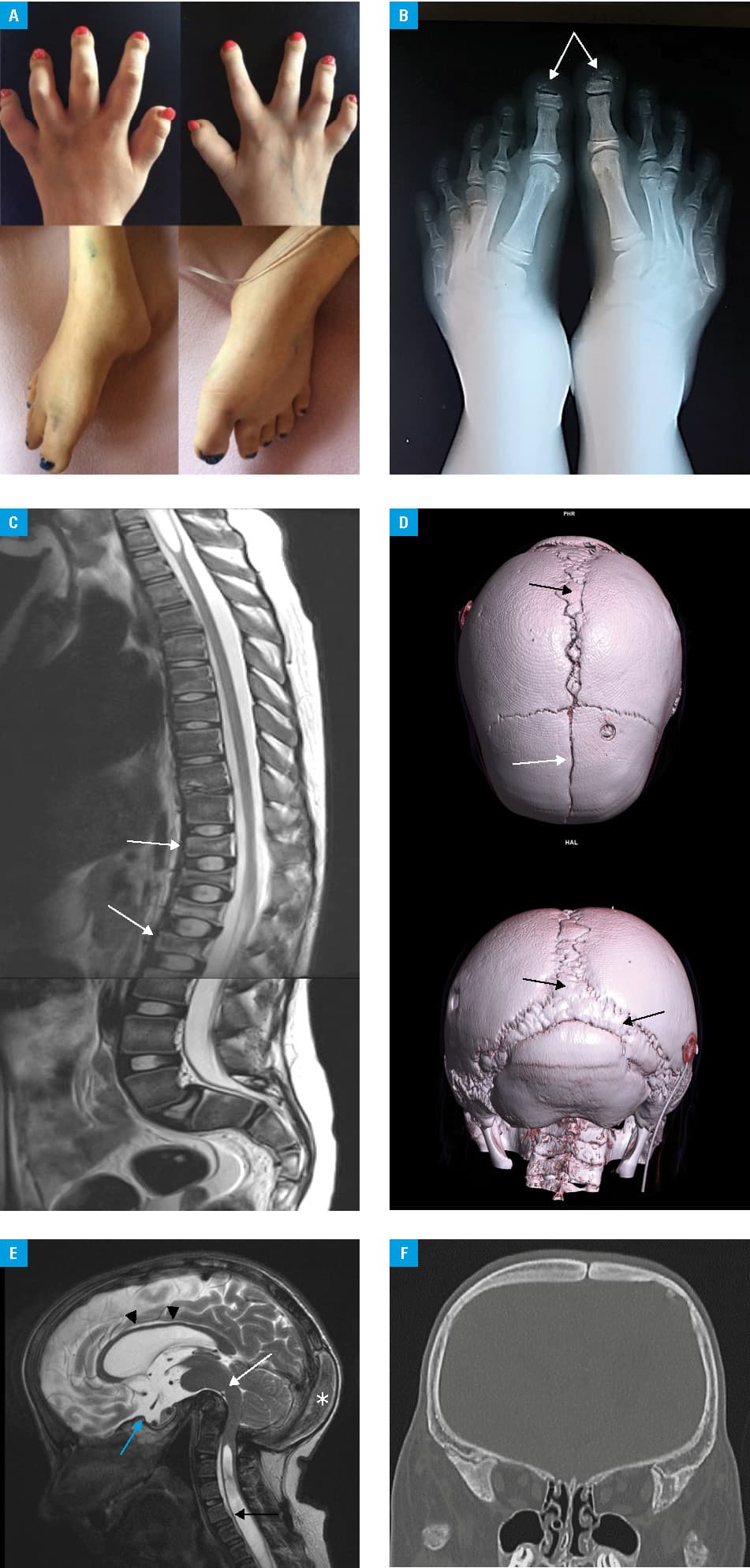
The patient presented with symptoms considered typical of HCS but their intensity seemed to be higher than in other cases described in the literature. To the best of our knowledge, the case described herein is the first report of the disease in Poland and was the result of a de novo mutation. A causative therapy does not exist for HCS, so the patients can only be treated symptomatically. Cranial base reconstruction was considered but due to high risk, the surgery was cancelled.
- Canalis E, Zanotti S. Hajdu‑Cheney syndrome: a review. Orphanet J Rare Dis. 2014; 9: 200. | Crossref
- Rodríguez L, Piqueras‑Sola B, Rodríguez‑Blanque R, et al. Hajdu‑Cheney syndrome: a systematic review of the literature. Int J Environ Res Public Health. 2020; 17: 6174. | Crossref
- Cheney WD. Acro‑osteolysis. Am J Roentgenol Radium Ther Nucl Med. 1965; 94: 595‑607.
- Hajdu N, Kauntze R. Cranio‑skeletal dysplasia. Br J Radiol. 1948; 21: 42‑48. | Crossref
- Brennan AM, Pauli RM. Hajdu‑Cheney syndrome: evolution of phenotype and clinical problems. Am J Med Genet. 2001; 100: 292‑310. | Crossref
ARTICLE INFORMATION