Pseudohypoparathyroidism type 1a caused by a GNAS gene mutation: over 40 years without a proper diagnosis



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Pseudohypoparathyroidism type 1a caused by a GNAS gene mutation: over 40 years without a proper diagnosis
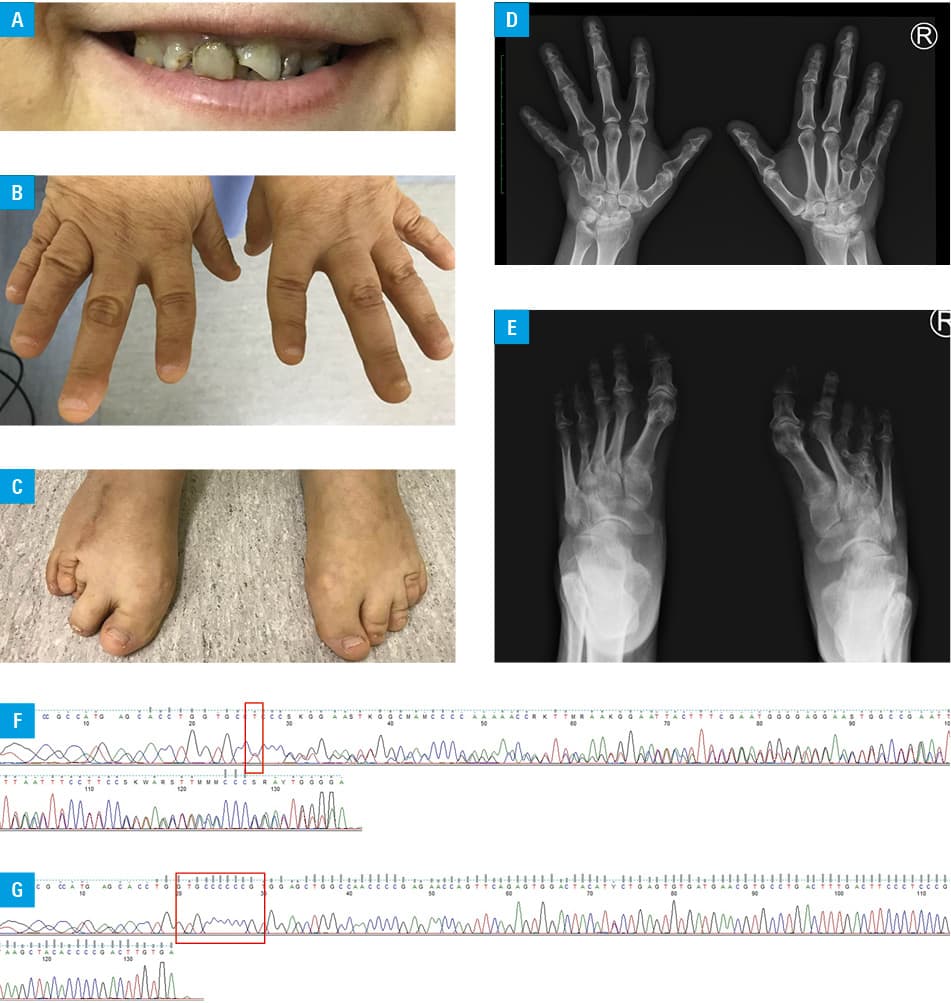
A 45‑year‑old woman was referred to the Endocrinology Clinic of Holy Cross Cancer Centre, Kielce, Poland due to hypercalcemia. She had a history of mild mental retardation, early‑onset obesity, orthopedic surgery for congenital skeletal defects of the right foot at 16 years of age, and cataract surgery on both eyes at 44 and 45 years of age. The patient had no siblings and has never been pregnant. At 42 years of age, hypocalcemia and hyperphosphatemia were detected in a regional endocrinology outpatient clinic without further diagnostics, and treatment with calcium carbonate (CaCO3) and alphacalcidol was commenced. At 45 years of age, she was admitted to a regional hospital due to renal insufficiency with an estimated glomerular filtration rate of 13.2 ml/min/1.73 m2. Hypercalcemia was also detected, but calcium supplementation was not modified. She had a short stature, round face, obesity (height, 1.44 m; weight, 64 kg), enamel hypoplasia, brachydactyly, and skeletal abnormalities of the feet (Figure 1A–1C). Additional investigations performed on index admission revealed hypercalcemia (3.39 mmol/l; reference range, 2.1–2.6 mmol/l), an elevated level of creatinine (2.4 mg/dl; reference range, 0.5–0.9 mg/dl), and a decreased level of parathormone (PTH) (12 pg/ml; reference range, 15.06–68.3 pg/ml). On X‑ray, bone anomalies typical of Albright hereditary osteodystrophy (AHO) were identified (Figure 1D and 1E). CaCO3 and alphacalcidol were discontinued. Renal parameters improved due to successful treatment of kidney disease, and then hypocalcemia reoccurred (2.06 mmol/l) along with an increase in the PTH level (158.8 pg/ml). CaCO3 and alphacalcidol were reintroduced. Her clinical presentation was suggestive of pseudohypoparathyroidism type 1a (PHP1a). However, her mother lacked a similar phenotype, despite the fact that PHP1a is caused by a maternally inherited mutation of the GNAS gene. Whole genome sequencing and Sanger sequencing detected the c.344_345insT p.(Val117ArgfsTer23) mutation in exon 5 of GNAS in the patient, but this genetic alteration was not identified in the patient’s mother (Figure 1F and 1G).
Pseudohypoparathyroidism is a rare genetic disorder characterized by resistance to PTH.1,2 There are several variants of PHP and their clinical characteristics depend on the type of genetic defect; mutations in the GNAS gene are the most frequent cause of PHP, while mutations in the PRKAR1A, PDE4D, and PDE3A genes are less common.1,2 In addition, the clinical presentation of PHP related to GNAS depends on the type of genetic defect (mutation / methylation defect), localization of mutation (eg, maternal / paternal allele), as well as paternal imprinting of GNAS in many organs, which may cause pseudo‑PHP with AHO, but without PTH resistance.1,2 In PHP, resistance to PTH results to a characteristic phenotype in the skeletal system called AHO which is typically observed in PHP1a, but not in PHP type 1b.2 In the kidneys, PTH‑dependent formation of 1,25‑dihydroxyvitamin D is impaired, leading to chronic hypocalcemia and hyperphosphatemia with secondary elevation of PTH levels.1-3 Nevertheless, in PHP, numerous early‑onset symptoms and signs related to congenital and chronic hypocalcemia may occur, which can facilitate recognition of this disorder.
Multiple mutations of GNAS have been reported to cause PHP1a. They are maternally inherited in 50% of cases, whereas the other 50% are de novo mutations.4 The presence of genetic alteration in our patient may be considered a de novo mutation in the GNAS gene; however, for each identified de novo pathogenic lesion, paternal germline mosaicism should be also considered.1,4 To date, there was only one published report of a GNAS mutation in the same location in a patient with PHP1a.5 In conclusion, due to the rarity of PHP, its proper diagnosis may be delayed for many years, despite the characteristic inherited phenotype and / or progressive complications of chronic hypocalcemia, especially in patients with no known family history of this disorder.
- Mantovani G, Bastepe M, Monk D, et al. Diagnosis and management of pseudohypoparathyroidism and related disorders: first international consensus statement. Nat Rev Endocrinol. 2018; 14: 476‑500. | Crossref
- Linglart A, Levine MA, Jüppner H. Pseudohypoparathyroidism. Endocrinol Metab Clin North Am. 2018; 47: 865‑888. | Crossref
- Shoback DM, Bilezikian JP, Costa AG, et al. Presentation of hypoparathyroidism: etiologies and clinical features. J Clin Endocrinol Metab. 2016; 101: 2300‑2312. | Crossref
- Kotanidou EP, Tsinopoulou V‑R, Serbis A, et al. Pseudohypoparathyroidism type 1A with normocalcaemia, due to the novel C.389A>G variant of exon 5 of the guanine nucleotide‑binding protein, α-stimulating gene. J Bone Metab. 2021; 28: 85‑89. | Crossref
- Park CH, Park HD, Lee SY, et al. Clinical, biochemical, and genetic analysis of Korean patients with pseudohypoparathyroidism type Ia. Ann Clin Lab Sci. 2010; 40: 261‑266.
ARTICLE INFORMATION