Genetically confirmed transthyretin amyloidosis primarily diagnosed as hypertrophic cardiomyopathy



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Genetically confirmed transthyretin amyloidosis primarily diagnosed as hypertrophic cardiomyopathy
Verification of echocardiographic appearances of hypertrophic cardiomyopathy (HCM) poses a diagnostic challenge. In some cases, after considering differential diagnoses, a proper diagnosis of transthyretin amyloidosis (ATTR) is particularly important due to the possibility of causative therapy. In ATTR, left ventricular hypertrophy is generated by amyloid fibrils that infiltrate the myocardium. Apart from diagnostic tools, multidisciplinary medical knowledge is required to solve the puzzle of extracardiac symptoms signaling ATTR.
This case involved a 60‑year‑old man from southern Poland. He was referred to our hospital with signs of heart failure (New York Heart Association class II), suspected as HCM, and gastrointestinal symptoms (frequent diarrhea defined as a separate disease). The patient’s detailed family history (only an oral report without medical records) revealed that his father had died of multiple myeloma and his cousin had died of amyloidosis (no information available about ATTR type), at the age of 59. The patient also disclosed extracardiac symptoms of carpal tunnel syndrome, which is characteristic of amyloidosis1. Left ventricular (LV) hypertrophy on electrocardiography was also observed as a typical sign of HCM (low voltage QRS complexes are common in amyloidosis).
Based on echocardiography, the patient demonstrated hypertrophy of LV, septal (2.1 cm during diastole), and posterior walls (1.6 cm during diastole). Interestingly, a myocardial noncompaction area was detected near the apex (Figure 1A). There have been only a few cases of HCM with noncompaction reported. The LV ejection fraction was moderately reduced (45%), and a nonrestrictive filling pattern was observed on Doppler examination. The global longitudinal strain (GLS) was abnormal, and there was “apical sparing” in the apical segment, which is a typical pattern of ATTR (Figure 1B). A myocardial noncompaction abnormality, however, may influence the apical sparing occurrence. Using echocardiographic criteria proposed by Jenni et al,2 we verified a myocardial noncompaction abnormality. The first criterion, a 2‑layer structure, was detected with a compacted thin epicardial band and a much thicker noncompacted endocardial layer of trabecular meshwork with deep endomyocardial spaces. The second diagnostic criterion was a maximal end systolic ratio of a noncompacted to compacted layer above 2. Finally, there was a color Doppler evidence of deep, perfused intertrabecular recesses. Myocardial overload was confirmed by a significant elevation of cardiac biomarkers: N‑terminal pro–B‑type natriuretic peptide (1887 pg/ml [reference range <125 pg/ml]) and high‑sensitivity troponin I (92 ng/ml [reference range <47.3 ng/ml]). Cardiac magnetic resonance imaging (MRI) confirmed the LV hypertrophy with myocardial noncompaction (Figure 1C), without late gadolinium enhancement. Verification of multiple myeloma was negative, with normal levels of kappa and lambda light chains. A small amyloid deposit was detected in a biopsy of the subcutaneous tissue.
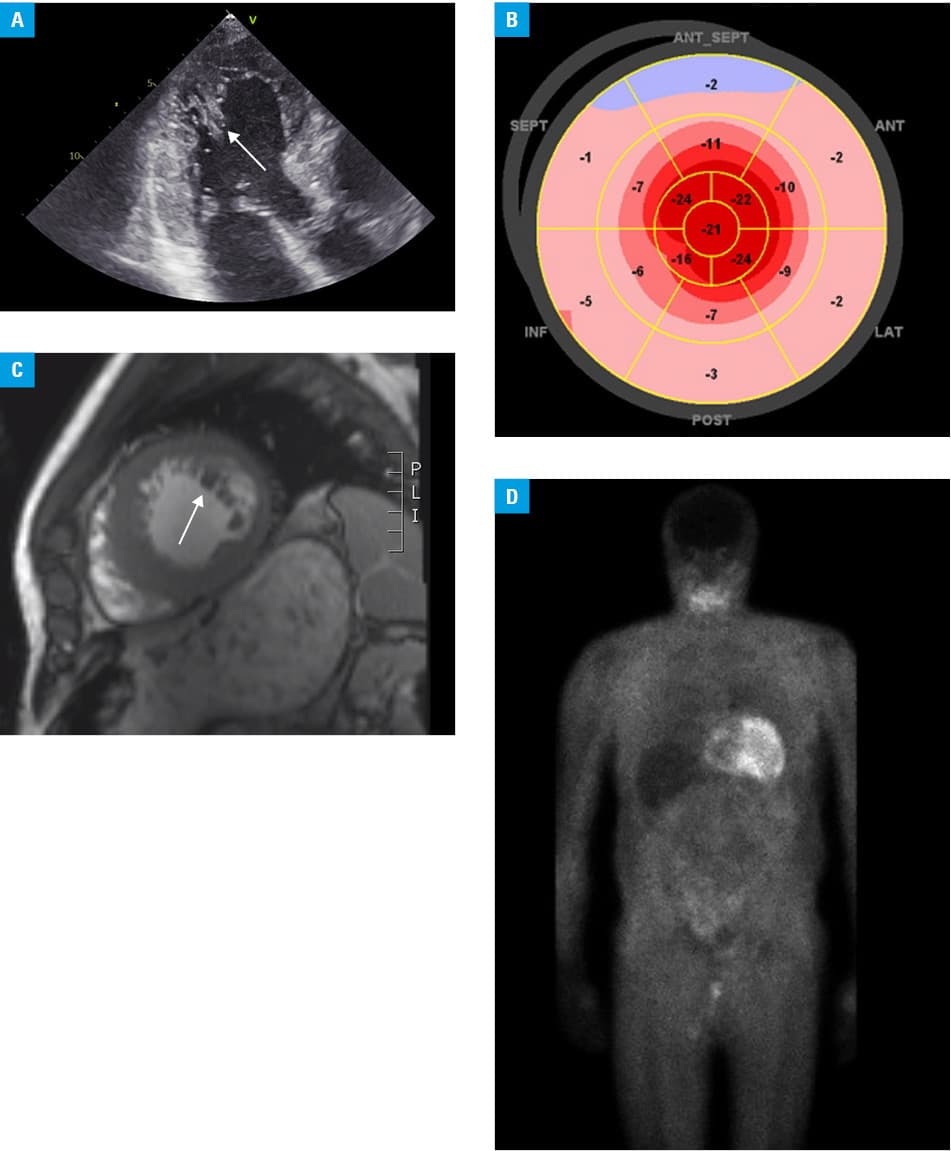
We performed 99mtechnetium‑labeled 3,3‑diphosphono‑1,2‑propanodicarboxylic acid scintigraphy. The examination revealed the radiotracer uptake in the heart (Perugini grade 3), a typical feature of ATTR (Figure 1D). A neurological examination revealed axonal polyneuropathy, which is common in the mutation variant. Utilizing genetic analysis, we recognized the ATTR variant c.157T>C (p.Phe53Leu), which is defined in the ClinVar NCBI database as pathogenic. Interestingly, this mutation is a candidate for an endemic one in southern Poland, as it was detected in 7 out of 10 patients in a recent study.3 We currently treat 8 patients with this mutation, which brings the total number of cases in Poland to 15. In a publication from another Polish center,4 the authors studied genetic variants in ATTR and described its heterogeneity. This is typical of nonendemic areas but the Phe53Leu variant was the most frequent.The patient described here has 3 daughters. The youngest one inherited the gene from her father, variant c.157T> C (p. Phe53Leu). The other daughters did not consent to further diagnostics. The patient is currently on low‑dose angiotensin‑converting enzyme inhibitor, a β-blocker, and an aldosterone antagonist. Due to a very high cost of the treatment, we took steps to provide the patient with tafamidis as part of a charitable pharmacy service.
- Petkow‑Dimitrow P, Rajtar‑Salwa R, Holcman K, et al. From hypertrophic cardiomyopathy to transthyretin amyloidosis: an unusual case and challenging diagnosis. Pol Arch Intern Med. 2020; 130: 153‑154. | Crossref
- Jenni R, Oechslin E, Schneider J, et al. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non‑compaction: a step towards classification as a distinct cardiomyopathy. Heart. 2001; 86: 666‑671. | Crossref
- Gawor M, Holcman K, Franaszczyk M, et al. Spectrum of transthyretin gene mutations and clinical characteristics of Polish patients with cardiac transthyretin amyloidosis. Cardiol J. 2020 Aug 11. [Epub ahead of print]. | Crossref
- Lipowska M, Drac H, Rowczenio D, et al. Transthyretin‑related familial amyloid polyneuropathy (ATTR‑FAP) in Poland – genetic and clinical presentation. Neurol Neurochir Pol. 2020; 54: 552‑560. | Crossref
ARTICLE INFORMATION