Hyperprolactinemia is a common hormonal disturbance associated with various etiologies. In the case of sellar tumors, elevated prolactin levels are most often due to active secretion by a prolactinoma. They may also result from an indirect effect of the tumor on the pituitary stalk.1
The skull base and sellar area are uncommon locations for giant cell tumors (GCTs), locally aggressive osteolytic neoplasms.2 Despite being histologically benign, the lesions present local recurrences and sporadically demonstrate metastatic potential. The most optimal therapeutic management of GCTs includes total surgical resection of the lesion.3 In the case of subtotal resections, a recurrence rate of over 50% was observed.4 The human monoclonal antibody against the receptor activator of nuclear factor kappa-Β ligand, denosumab, is an approved agent for the treatment of unresectable tumors.5
A 26‑year‑old woman presented with secondary amenorrhea for 4 months. Initially, she was diagnosed by a gynecologist, and pregnancy was ruled out. The hormonal assessment showed a moderately elevated serum prolactin level (49.97 ng/ml; reference range, 4.79–23.3 ng/ml), while other hormone levels were normal. Magnetic resonance imaging (MRI) of the head revealed a posterior sellar mass without suprasellar extension (1.0 × 1.4 × 1.3 cm) (Figure 1A). Based on imaging studies and clinical manifestations, a diagnosis of macroadenoma was made. Due to the absence of visual symptoms and no signs of hypopituitarism or active hormonal secretion, treatment with bromocriptine at a dose of 2.5 mg twice daily was recommended, which led to restoration of the menstrual cycle.
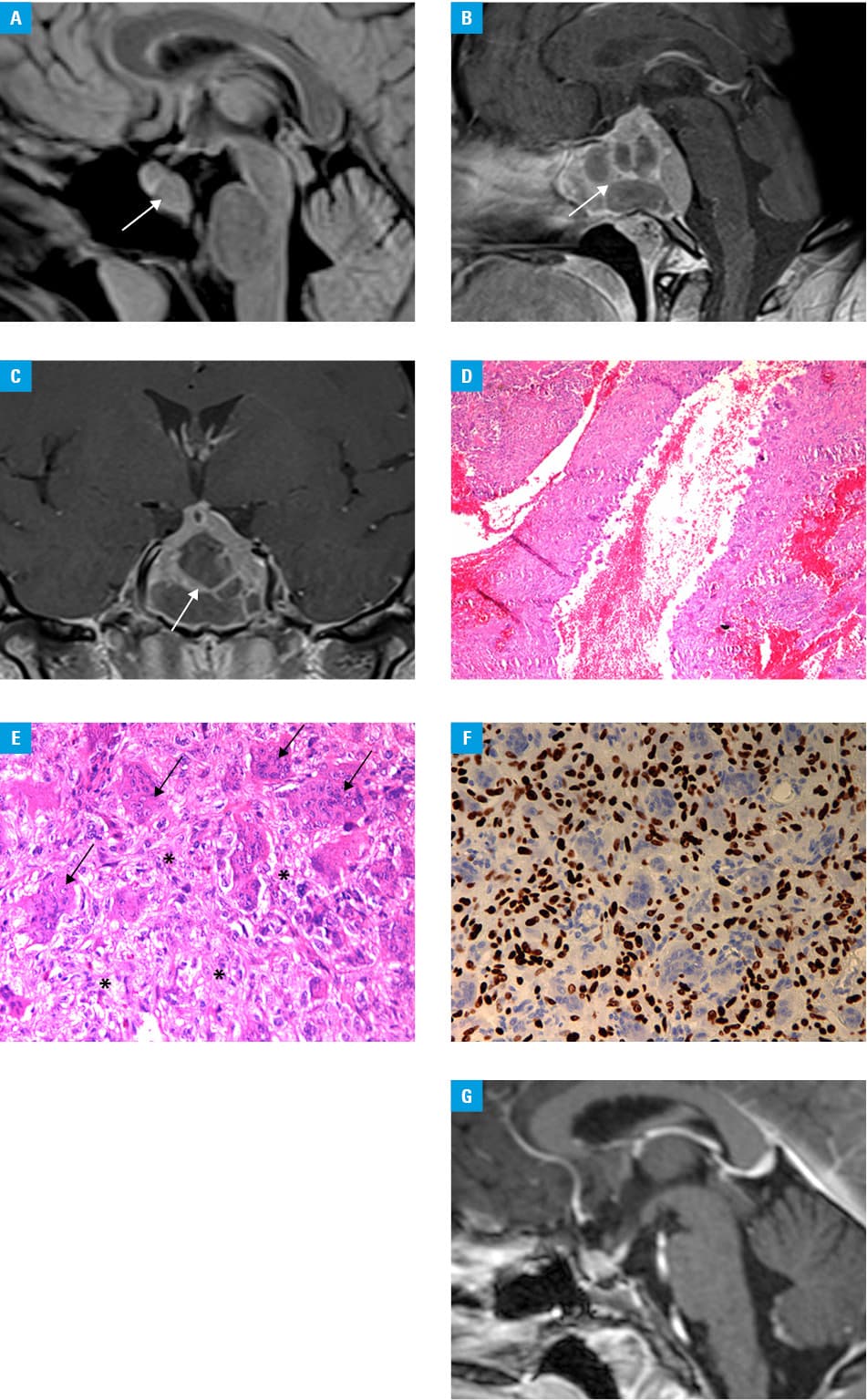
After 5 months, the patient presented with severe headaches with orbital pain accompanied by double vision. The second MRI revealed a giant solid‑cystic tumor of the clivus, sella, and sphenoid sinus (4.6 × 3.8 × 4.0 cm) with brainstem compression and infiltration of the left cavernous sinus (Figure 1B and 1C; Supplementary material, Figures S1–S4). Computed tomography showed an osteolytic hypodense tumor with mild vascular enhancement of its solid part (Supplementary material, Figures S1 and S2). On neurological examination, left abducens nerve paresis and asymmetrical bitemporal visual field defects were observed.
An endoscopic transnasal transsphenoidal near total resection of the tumor was performed. A small residual tumor was left in the cavernous sinus to reduce the risk of cranial nerve deficits and internal carotid artery injury. Histopathological examination revealed a GCT with a secondary aneurysmal bone cyst (Figure 1D and 1E).
The H3‑3A gene mutation (formerly H3F3A) was detected using antihistone H3.3 G34W rabbit monoclonal antibody, and was confirmed by direct sequencing of exon 2 of the H3‑3A gene, thus fulfilling the 2020 World Health Organization criteria for the diagnosis of GCT (Figure 1F).
The postoperative course was uneventful. Headache, diplopia, and visual impairment subsided. Pituitary hormonal status after the surgery was normal (prolactin level of 7.0 ng/ml, off pharmacotherapy). Due to the subtotal resection, therapy with denosumab was initiated at a dose of 120 mg subcutaneously every 28–42 days. Over a 2‑year follow‑up, there was no progression of the small residual GCT (Figure 1G).
Hyperprolactinemia is a common hormonal disturbance in the course of pituitary and parasellar tumors. Our case showed an extremely rare type of tumor in this location with a complex clinical presentation. The rapid clinical and radiological progression of the tumor despite typical pharmacological treatment of hyperprolactinemia should prompt a revision of diagnosis. Pituitary macroadenoma associated with only moderate rather than marked hyperprolactinemia requires a careful differential diagnosis.
- Samperi I, Lithgow K, Karavitaki N. Hyperprolactinaemia. J Clin Med. 2019; 8: 2203. | Crossref
- Shibao S, Toda M, Yoshida K. Giant cell tumors of the clivus: case report and literature review. Surg Neurol Int. 2015; 6: S623‑S627. | Crossref
- Huh A, Villelli N, Martinez D, et al. Denosumab treatment for a residual giant cell tumor of the clivus: a case report and review of the literature. World Neurosurg. 2018; 118: 98‑110. | Crossref
- Xará-Leite F, Coutinho L, Fleming C, et al. Can denosumab cure giant cell tumors of the spine? A case report and literature review. Eur J Orthop Surg Traumatol. 2020; 30: 19‑23. | Crossref
- Goto Y, Furuno Y, Kawabe T, et al. Treatment of a skull‑base giant cell tumor with endoscopic endonasal resection and denosumab: case report. J Neurosurg. 2017; 126: 431‑434. | Crossref
SUPPLEMENTARY MATERIAL
ARTICLE INFORMATION