Statins in prevention of thromboembolic events: from seminal studies to recent advances
Key words: blood coagulation, statins, thrombin generation, thromboembolism, tissue factor



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Statins in prevention of thromboembolic events: from seminal studies to recent advances
Statins, 3‑hydroxy‑methylglutaryl coenzyme A reductase inhibitors, reduce the rate of cardiovascular events in primary and secondary prevention, and represent a cornerstone in the management of atherosclerotic vascular disease. Statins were also shown to be effective in lowering the risk of venous thromboembolism (VTE) in its primary and secondary prevention, but their use in VTE prophylaxis is still not approved and recommended in current guidelines. Several antithrombotic mechanisms of these cholesterol‑lowering agents, largely independent of the magnitude of low‑density lipoprotein cholesterol reduction, were demonstrated in a broad spectrum of experimental in vitro and in vivo models. However, these studies yielded inconsistent results, such as substantial differences related to the use of specific statins, their dose or final concentration, and even the clinical setting (patients with cardiovascular disease or VTE vs healthy subjects). Anticoagulant properties of statins, reported for the first time 25 years ago, involve downregulation of tissue factor expression with a subsequent decreased thrombin generation and inhibition of thrombin‑mediated reactions, including factor V and factor XIII activation, and enhanced endothelial thrombomodulin expression resulting in increased protein C activation. Enhanced fibrinolysis was also reported partly as a result of reduced activity of fibrinolysis inhibitors such as plasminogen activator inhibitor‑1 (PAI‑1) and thrombin‑activatable fibrinolysis inhibitor. This review summarizes the findings of the studies from the 1990s until the most recent reports to update our knowledge on the impact of statins on blood coagulation and its potential clinical relevance.
Introduction
Statins (3‑hydroxy‑methylglutaryl coenzyme A [HMG‑CoA] reductase inhibitors) are highly effective in reducing low‑density lipoprotein (LDL) cholesterol (LDL‑C). At typical daily doses statins reduce total and LDL cholesterol by 18%–60% with a decrease in triglycerides by 10%–30%, and a slight increase in high‑density lipoprotein (HDL) cholesterol (8%).1 Over 20 years ago, statins, apart from cholesterol lowering, were postulated to exert so‑called pleiotropic, largely beneficial effects that may have contributed to more favorable outcomes observed in clinical trials on their use in cardiovascular disease.1 It was suggested that as these drugs decrease the synthesis of isoprenoid intermediates (eg, farnesyl pyrophosphate and geranylgeranyl pyrophosphate [GGPP]) and alter posttranslational protein prenylation, which might affect, among others, cell proliferation and signaling pathways,2 changes in the expression of multiple proteins, largely via the inhibition of Rho, Ras, and Rac, represent additional effects of statins.
Clinical and experimental studies show that the pleiotropic effects involve reduced atherosclerotic plaque progression,3 plaque regression4,5 and stabilization,6 anti‑inflammatory effects,7,8 reduction of myocardial ischemia / reperfusion injury,9 and antiatherogenic response.6,10
Antithrombotic effects of statins involving endothelial cells, platelets, and coagulation proteins along with fibrinolysis proteins were reported in diverse experimental models.11,12
This review focuses on the effect of statins on blood coagulation and its potential relevance for the prevention of venous thromboembolism (VTE) based on studies published over the last 25 years.
Statins and prevention of thromboembolism: clinical aspects
Data from the Cholesterol Treatment Trialists’ Collaboration indicated that statins lowered the risk of major vascular events by 22%.13 Lowering LDL‑C levels with statins during primary and secondary prevention reduced the risk of coronary artery disease (CAD) events and death.14 The current European and United States dyslipidemia management guidelines recommend identifying patients at high cardiovascular risk and treating them to achieve low LDL‑C levels. The European guidelines set the goal of below 1.4 mmol/l (<55 mg/dl) in the patients with high‑risk cardiovascular disease. Despite a growing role of proprotein convertase subtilisin / kexin type 9 inhibitors, for many high‑risk patients who cannot achieve LDL‑C goals with statins or ezetimibe, HMG‑CoA inhibitors are still the cornerstone of therapy.15,16
In addition to well‑known benefits of statins in long‑term secondary prevention of CAD, statin therapy significantly reduces the risk of recurrent cardiovascular events during and after the acute coronary syndrome (ACS).17 A meta‑analysis of randomized controlled trials by Hulten et al17 demonstrated that early, intensive statin therapy of ACS decreased the mortality and cardiovascular events rate in a 2‑year follow‑up (hazard ratio [HR], 0.81; 95% CI, 0.77–0.87; P<0.001). Significant survival benefit was observed after 12 months since the ACS.17 However, statins initiated within 14 days following ACS did not improve hard clinical outcomes such as death or myocardial infarction (MI),18 as risk ratio for a combined end point of death, MI, and stroke of early statin therapy vs control was 0.93 (95% CI, 0.80–1.08) at 1 month and 0.93 (95% CI, 0.81–1.06) at 4 months following ACS. Early initiation of a high‑dose statin therapy in ACS was shown to reduce thrombotic events within the first 4 months.19
Ray et al20 reported for the first time in 2001 that statins, regardless of the dosage regimen, reduce VTE risk by 22%. This observation was supported by the results of the Heart and Estrogen / progestin Replacement Study (HERS), in which even larger (about 50%) reduction in VTE risk was found among statin recipients.21
Cumulative analyses demonstrated statins as capable of decreasing VTE risk, suggesting their class effect, regardless of the patient’s age and presence of the cardiovascular disease.22 A meta‑analysis of observational studies demonstrated that statin administration is associated with lower VTE risk (odds ratio [OR], 0.67; 95% CI, 0.53–0.84), mainly due to a lowered risk of deep vein thrombosis (DVT) (OR, 0.53; 95% CI, 0.22–1.29).23 A meta‑analysis of 36 studies revealed a beneficial effect of statins on VTE in both observational and interventional studies.24 Another meta‑analysis of 7 observational studies published before 201725 confirmed that statins significantly lower the recurrent VTE rate (relative risk [RR], 0.73; 95% CI, 0.68–0.79), as compared with non‑statin control, with similar reductions for recurrent pulmonary embolism (PE) (RR, 0.75; 95% CI, 0.58–0.96) and DVT (RR, 0.66; 95% CI, 0.60–0.71).
From a methodological perspective, the most compelling evidence for statin‑induced decrease of VTE risk came from the JUPITER (Justification for the Use of statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) study in 2009.26 Glynn et al26 demonstrated that in asymptomatic men ≥ 50 or women ≥ 60 years, with LDL‑C below 3.4 mmol/l and CRP above 2 mg/l, rosuvastatin 20 mg/d markedly lowered VTE risk, as compared with placebo, during a 1.9‑year follow‑up (HR, 0.57; 95% CI, 0.37–0.86). The most pronounced decrease (by 55%) was observed for isolated DVT, without differences between unprovoked (39%) and provoked VTE (48%).
Lowered risk of thrombosis in statin recipients was reported in high‑risk populations including those with active cancer (OR, 0.33; 95% CI, 0.19–0.57)27 and nephrotic syndrome (HR, 0.2; 95% CI, 0.1–0.7).28 A Dutch, population‑based registry of pharmacy records for patients hospitalized with an acute episode of PE showed that treatment with statins was associated with a reduced risk of recurrent PE (HR, 0.50; 95% CI, 0.36–0.70), both during and after withdrawal of oral anticoagulants.29In a huge meta‑analysis involving 22 trials of statin vs control and 7 trials of an intensive vs a standard‑dose statin regimen published by Rahimi et al30 in 2012, statins failed to reduce VTE occurrence and only showed a trend toward such a reduction (OR, 0.89; 95% CI, 0.78–1.01; P=0.08). Among Canadian patients aged 65 or over,31 with a history of VTE, those taking statins were less likely to experience VTE recurrence (HR, 0.74; 95% CI, 0.61–0.89) during a mean follow‑up of 3 years. The risk reduction was greater in individuals receiving these drugs for longer than 12 months. Additional data on the elderly (mean age 75) were published by Kronenberg et al,32 who concluded that in acute VTE patients statins reduced the risk of recurrent episodes to a much greater degree (80%) than previously estimated.
Statins do not increase bleeding risk. A post‑hoc analysis of a randomized trial in patients with cerebrovascular disease showed a higher rate of hemorrhagic stroke in individuals on high‑dose atorvastatin as compared with placebo (HR, 1.66; 95% CI, 1.08–2.55),33 however, a meta‑analysis of randomized controlled statin trials failed to demonstrate the increased bleeding risk associated with this class of hypolipemic agents.34
Statins and blood clotting: old and new mechanisms
Numerous in vitro studies indicated that downregulation of tissue factor (TF) expression can be observed in response to statins. In 1997, simvastatin and fluvastatin, but not pravastatin, were reported to reduce TF expression and activity in lipopolysaccharide (LPS)-stimulated human monocytes / macrophages.35 This reduction was reversed by mevalonate or GGPP, with a significant contribution of inhibition of transcription nuclear factor kappa B activation. A 3‑fold reduction in TF expression and activity was observed in human LPS‑stimulated monocytes isolated from hypercholesterolemic participants treated with simvastatin 20 mg/d.36 The same statin was shown to inhibit thrombin‑induced TF expression in human aortic endothelial cells and aortic smooth muscle cells.37 The inhibition of Rho‑kinase‑dependent Akt dephosphorylation was involved in downregulation of endothelial TF expression by statins.37,38 Later on, it was demonstrated that fluvastatin and rosuvastatin can block this mechanism of inducing TF expression by inhibiting ERK1/2 phosphorylation and protease activated receptor‑1 relocation in a pathway independent of isoprenoids.39 Downregulation of TF associated with suppressed inflammation but not with cholesterol levels, was documented for various statins in animal models, such as apolipoprotein E–deficient mice or cholesterol‑fed rabbits.40-42 In human patients, 4–6 months of treatment with atorvastatin resulted in 29% lower TF antigen levels and 56% lower TF activity in atherosclerotic plaques removed during endarterectomy as compared with the values for subjects receiving placebo.43 Statin‑induced decrease in TF expression was reported in atherosclerotic plaques obtained from the coronary artery.44 Simvastatin, assessed in LDL receptor‑deficient mice on a 4‑week high‑fat diet, was found to decrease oxidized LDL (oxLDL) levels and reduce white blood cell TF expression, while the number of TF‑positive exosomes and plasma thrombin markers were unchanged. Interestingly, TF expression in hypercholesterolemic mice and monkeys was mediated by the monocyte CD36 / TLR4 / TLR6 heterotrimeric complex enhanced by oxLDL, and simvastatin lowered TF and interleukin‑6 expression by lowering oxLDL levels.45
Despite the fact that downregulation of TF, the main initiator of blood coagulation in vivo, should imply suppression of thrombin formation, studies on the impact of statins on thrombin generation yielded inconsistent results over the last 25 years. Plasma thrombin markers were not significantly lower in patients on statins or this effect was minimal.46-48 Platelet- or monocyte‑based models were used to investigate suppressed thrombin generation in response to statins. Pravastatin was reported to reduce platelet‑dependent thrombin generation in patients with hypercholesterolemia.49 Ferro et al50 described a marked inhibition of thrombin generation in plasma incubated with mononuclear cells obtained from healthy individuals and hypercholesterolemic patients receiving simvastatin. Although the formation of baseline prothrombin fragments 1.2 (F1.2) was higher in hypercholesterolemic patients, the simvastatin‑induced reduction in thrombin generation was similar in both groups regardless of blood cholesterol levels. This reduction varied from below 10% at simvastatin concentration of 0.01 pmol/l to 55% at the drug concentration of 10 pmol/l.50 However, Lindhout et al51 failed to confirm the inhibition of phosphatidylserine‑dependent prothrombinase activity in the platelets of healthy individuals receiving simvastatin 40 mg/d for 2 weeks.
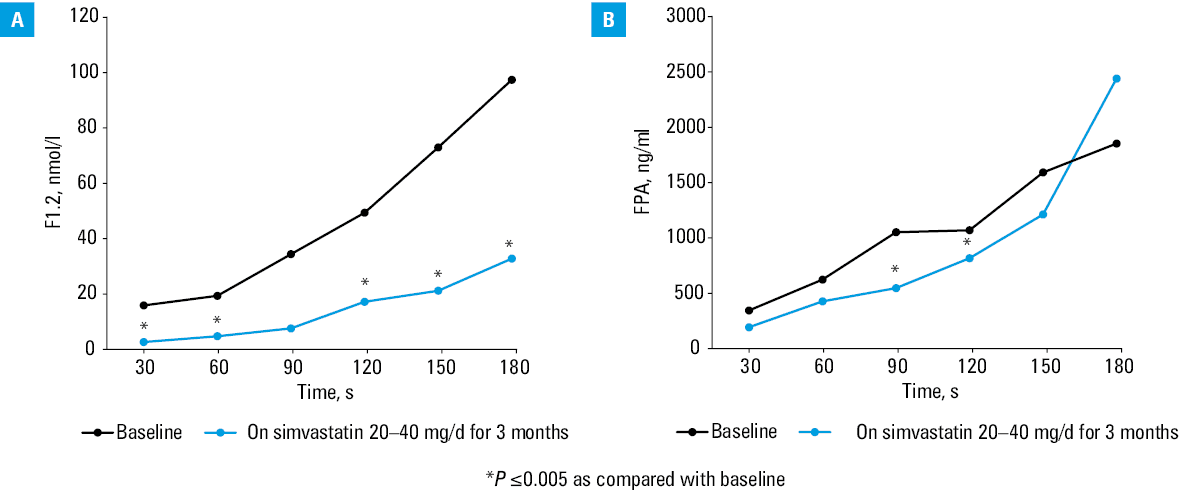
The group of Professor A. Szczeklik from Kraków, Poland, largely contributed to the elucidation of statin‑mediated modulation of TF‑initiated thrombin generation in humans. His team modified a model of microvascular injury in the 1980s,52 and introduced measurements of several thrombosis markers in the supernatant of blood samples obtained from standardized bleeding‑time wounds on a forearm, at the sites of hemostatic plug formation performed with various bleeding‑time devices. Thrombin markers, such as F1.2, fibrinopeptide A, and thrombin‑antithrombin complexes (TAT), were used to assess the kinetics of the blood coagulation activation before and during statin therapy. The first study from the Szczeklik’s group was published in J Am Coll Cardiol in 1999.53 The study assessed patients with hypercholesterolemia, defined as total cholesterol above 6.5 mmol/l, in whom a 3‑month treatment with simvastatin 20–40 mg/d, reduced the maximum rate and level of thrombin generation at the site of microvascular injury53 (Figure 1). Hypercholesterolemic patients with CAD, defined as total cholesterol between 5.2 and 6.5 mmol/l, exhibited also slower rate of thrombin formation after simvastatin administration.54 A. Szczeklik and K. G. Mann implemented Western blot technique to provide a more refined evaluation of changes in coagulation proteins and proteins detectable in blood oozing from bleeding time skin incisions performed in statin users. In a paper published in Circulation in 2001, it was demonstrated that in hypercholesterolemic subjects, simvastatin 20–40 mg/d administered for 3 months led to a decrease in the rate of prothrombin depletion and prothrombin activation, respectively, without any association with lipid profiles55 (Figure 2A). The subsequent changes in additional reactions involved reduced factor Va generation by 19% and by 20% slower thrombin‑induced FXIII activation.55 In high CAD risk patients with LDL‑C above 3.4 mmol/l, simvastatin 40 mg/d lowered plasma TAT and total thrombin generated at the site of microvascular injury by 30% as soon as after 3 days, and independently of cholesterol reduction.56 Similar effects were observed in CAD patients following 1 month of atorvastatin (40 mg/d), with rates of thrombin formation reduced by 34%–40% and thrombin‑induced FV activation by 22%.57 In hypercholesterolemic patients atorvastatin 10 mg/d given for 3 days decreased plasma F1.2.58 Similarly, simvastatin 40 mg/d administered for 3 days in subjects with LDL‑C above 3.4 mmol/l caused a 30% decrease in thrombin generation rates assessed at the site of hemostatic plug formation, along with depressed thrombin‑induced factor V activation and enhanced factor Va inactivation59 (Figure 2B).
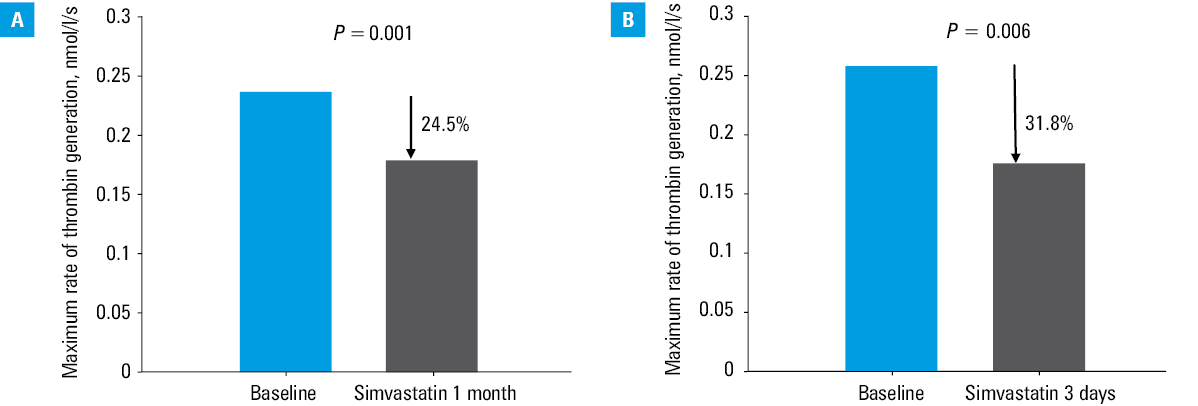
ACS studies failed to confirm reduced thrombin generation in circulating blood even at high‑dose atorvastatin.60 Pastuszczak et al61 reported that statin administration for at least a month prior to ACS can reduce thrombin generation and platelet activation assessed at the site of vascular injury in patients assessed within the first 12 hours. These results imply that while statins may be too weak to overcome potent prothrombotic mechanisms, they may favorably modulate thrombogenicity in ACS.
Genetic factors affecting statin‑induced modulation of blood coagulation were suggested by a study in which high‑risk individuals carrying +5466AG TF allele (~16% of the European population), experienced a greater decrease in thrombin formation rate following simvastatin use than those with +5466AA genotype.62
Using another common methodological approach, that is, a calibrated automated thrombogram, Dutch investigators in a randomized, controlled trial (START, STAtins Reduce Thrombophilia) confirmed that rosuvastatin 20 mg/d administered for 1 month caused a relative reduction in thrombin generation as evidenced by 10.4% lower endogenous thrombin potential and by 5% lower peak thrombin concentration, which were the most pronounced in patients with unprovoked VTE and in those with cardiovascular risk factors.63 To further elucidate the mechanisms underlying the anticoagulant effects of statins, the same group determined prothrombotic phospholipid activity with a FXa‑dependent clotting assay before and after 28 days of rosuvastatin treatment, and observed a 22% reduction in this activity, with the greatest effect in patients with prior PE.64
The reported effects of statins on fibrinogen concentrations include their increase, decrease, or most often, no significant change.65-67 Given the fact that changes in fibrinogen concentrations strongly affect fibrin clot formation and its properties, there is evidence that statin‑associated alterations to fibrin clot structure and / or functions, especially its susceptibility to plasmin‑mediated lysis, are related to changes in fibrin bound proteins or posttranslational modifications at similar plasma fibrinogen levels in patients receiving statins.
Most statins, except for pravastatin, demonstrated downregulation of plasminogen activator inhibitor‑1 (PAI‑1) expression associated with increased tissue‑type plasminogen activator (t‑PA) expression in monocytes, smooth muscle cells, and endothelial cells,68-70 which might have enhanced fibrinolytic capacity. Downregulation of PAI‑1 expression as a result of statin administration was reported to depend on the inhibition of Rho family proteins and may involve activation of PI‑3 kinase / Akt signaling pathways.71 In addition, statins decrease thrombin‑activatable fibrinolysis inhibitor (TAFI) (carboxypeptidase U) levels.72,73 Recent studies, including those in murine models, confirmed reduced TAFI in hypercholesterolemia in response to statins.74,75 Statins were found to enhance fibrinolysis, as evidenced by lower levels of plasmin‑antiplasmin complexes in hypercholesterolemic patients. The patients receiving atorvastatin 10 mg/d exhibited a reduction in P‑selectin but not in total cholesterol.73
There is compelling evidence that the CAD, including ACS, is associated with prothrombotic fibrin clot characteristics, including formation of more compact fibrin clot structures composed of generally thinner fibers of impaired lysability.76 In 2006, we were the first to demonstrate that statins can favorably change the fibrin clot phenotype including the density of fiber networks and their susceptibility to lysis. Administration of simvastatin or atorvastatin 40 mg/d for 28 days led to a 20% greater fibrin clot permeability and faster clot lysis in individuals at high cardiovascular risk.77 Similar observations were made in patients without CAD, who had LDL‑C below 3.4 mmol/l and received simvastatin at up to 20 mg/d.78 Recently we observed that CAD patients, who achieved LDL‑C of 1.8 mmol/l or lower on high‑dose statin therapy, demonstrated a 30% increase in clot permeability and by 17% shorter clot lysis time. There were no similar changes in the patients with LDL‑C above 1.8 mmol/l despite treatment with atorvastatin 80 mg/d or rosuvastatin 40 mg/d (Undas A, unpublished data). The effect of statins on fibrin characteristics was also reported in patients with a history of VTE, as evidenced by increased plasma fibrin clot permeability (by 23%) and lysability (by 15%–20%), regardless of drug‑induced cholesterol reductions, following atorvastatin 40 mg/d for 3 days.79 A recent START study,80 employing a different methodology, confirmed that statins, administered as rosuvastatin 20 mg/d for 28 days, enhanced fibrinolytic potential in patients following VTE. This effect was not associated with any changes in PAI‑1, though TAFI levels were 5% lower following such a therapy.80
Thrombomodulin (TM)-related anticoagulant effects were attributed to statins based on experimental papers published mainly in the years 2000–2010. Despite negligible changes in plasma soluble TM concentrations during statin treatment,12 the impact of statins on endothelial TM expression was reported by several groups. In 2003, Masamura et al81 demonstrated that pitavastatin increased transcription of TM gene and endothelial TM expression through the inhibition of Rho protein, Rac / Cdc42, reversed by mevalonate or GGPP. An exposure to atorvastatin 10 pmol/l for 24 hours led to a 10‑fold increase in TM transcripts, accompanied by a 2- to 3‑fold increase in TM protein, cell surface TM antigen, and PC activation. Simvastatin and atorvastatin diminished tumor necrosis factor alpha (TNF‑a)–induced downregulation of endothelial TM expression in relation to increased nitric oxide (NO) generation.82 Moreover, it was shown that administration of simvastatin and lovastatin, in contrast to pravastatin, can enhance the expression of a transcription factor, Kruppel‑like factor 2 (KLF2), which results in increased TM expression.83 Rossi et al84 demonstrated higher KLF2 and TM expression, associated with changes in endothelial NO synthase expression, following a combination of simvastatin and low shear stress. Simvastatin blocked the suppression of endothelial TM and endothelial NO synthase expression induced by proinflammatory cytokines, irrespective of the shear stress. Rosuvastatin produced less pronounced, though similar, effects on endothelial cells.85 Fu et al86reported that statins induced heat shock transcription factor 1 dissociation from the heat stress protein‑90 and activation of KLF2 in cultured human umbilical vein and human coronary artery endothelial cells. Of note, before the publication by Masamura et al,81 we observed in 2001 that the treatment with simvastatin may accelerate activated protein C‑mediated FVa inactivation in a model of microvascular skin injury,55 with a similar effect reported as early as after 3 doses of this drug in 2005.59
Tissue factor pathway inhibitor (TFPI) was found to be decreased by statins without reduction in free TFPI in hyperlipidemic individuals or patients with CAD.47,87 However, negligible statin‑induced effects on TFPI levels were also reported.88
In the Multi‑Ethnic Study of Atherosclerosis (MESA), involving middle‑aged and elderly healthy individuals, statin administration caused a 3% decrease in FVIII as compared with no statin use.65 In the randomized START study, Biedermann et al89 reported that rosuvastatin 20 mg/d, administered for 4 weeks, lowered FVIII by about 6% in VTE patients, particularly those with unprovoked episodes or with CAD risk factors. This change was accompanied by a 4% reduction in FXI, a 3% reduction in D‑dimer, and 4% drop in FVII.89 A reduction in von Willebrand factor, which is a carrier of FVIII in the circulation, was reported in a meta‑analysis in 2016.90 Moon et al91 provided evidence that statins increased the hepatic expression of LDL receptor‑related protein, representing the family of LDL receptors, and thus contributing largely to FVIII clearance.
Lower plasma FXI levels were reported in statin users aged 45 to 65 years, with a history of thrombosis, as compared with those receiving other hypolipemic agents.92
Very recently, in Mendalian randomization perfomed on data from Japan, genetically mimicked effects of statins showed increased prothrombin time, but not activated partial thromboplastin time.93
Conclusion
There is evidence that statins, powerful cholesterol‑lowering drugs, may produce antithrombotic effects including those associated with reduced thrombin generation. Much effort was put in to clarify the mechanisms via which statins may exert such potentially beneficial effects, though their clinical relevance remains uncertain. Anticoagulant effects of statins observed as early as after 1–3 days of their administration are most evident in hypercholesterolemic patients, despite the fact that the majority of antithrombotic effects show no association with the magnitude of LDL‑C reduction or duration of treatment. Statins, when compared with oral anticoagulants, have a good safety profile and do not cause bleeding. Therefore, their use in such a common disease as VTE could provide huge clinical benefits, particularly in patients at high risk of bleeding, who require long‑term VTE prevention. It remains to be established with high‑quality evidence whether or not statins are effective in VTE prevention. A rather impressive amount of data from several research groups strongly support this hypothesis and further studies on this topic seem promising.
- Takemoto M, Liao JK. Pleiotropic effects of 3‑hydroxy‑3‑methylglutaryl coenzyme A reductase inhibitors. Arterioscler Thromb Vasc Biol. 2001; 21: 1712‑1719. | Crossref
- McTaggart SJ. Isoprenylated proteins. Cell Mol Life Sci. 2006; 63: 255‑267. | Crossref
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Effect of intensive compared with moderate lipid‑lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA. 2004; 291: 1071‑1080. | Crossref
- Nicholls SJ, Tuzcu EM, Sipahi I, et al. Statins, high‑density lipoprotein cholesterol, and regression of coronary atherosclerosis. JAMA. 2007; 297: 499‑508. | Crossref
- Nissen SE, Nicholls SJ, Sipahi I, et al. Effect of very high‑intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006; 295: 1556‑1565. | Crossref
ARTICLE INFORMATION