Molecular cytogenetics in acute myeloid leukemia in adult patients: practical implications
Key words: acute myeloid leukemia, chromosome abnormality, karyotype, mutation, prognostication



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Molecular cytogenetics in acute myeloid leukemia in adult patients: practical implications
Throughout the last 50 years, cytogenetic analyses of pretreatment bone marrow and / or blood samples from patients diagnosed with acute myeloid leukemia (AML) revealed a large number of recurring chromosome aberrations, both structural and numerical. Using standard banding methods and, more recently, molecular cytogenetic techniques, such as fluorescence in situ hybridization, spectral karyotyping, multiplex fluorescence in situ hybridization and comparative genomic hybridization, cytogenetic investigations detect acquired abnormalities that, together with submicroscopic gene mutations and changes in gene expression, strongly influence the clinical features and prognosis of patients with AML. Selected reciprocal translocations and inversions and their molecular counterparts, as well as a number of unbalanced chromosome abnormalities are used, together with bone marrow morphology, immunophenotype, and clinical characteristics, to define separate AML entities in the World Health Organization Classification of Haematolymphoid Tumours. Moreover, cytogenetic findings (and specific gene mutations) are being used in genetic risk classifications, such as the 2022 European LeukemiaNet classification. Such classifications divide patients into broad prognostic categories: favorable, intermediate, and adverse, which are useful in the management of adults with AML. In this article, I review the present data on recurrent chromosome rearrangements in AML and on correlations between cytogenetic findings and clinical features and treatment outcomes of adult patients diagnosed with AML.
Introduction
During the last 50 years, cytogenetic investigations of acute myeloid leukemia (AML), a neoplastic disease characterized by deregulation of proliferation, differentiation, and / or programmed cell death (apoptosis) of hematopoietic progenitor cells, brought about major advances in our understanding of the genetic basis of this disorder and its considerable histopathologic, immunophenotypic, and clinical heterogeneity. As early as in 1972, de la Chapelle et al1 reported a sole trisomy of chromosome 8, and in 1973, Janet D. Rowley2 described a reciprocal translocation t(8;21)(q22;q22), which are, respectively, numerical and structural abnormalities that were later shown to be among the most frequent and clinically relevant recurrent chromosome aberrations in AML.3,4 To date, over 300 recurrent chromosome abnormalities have been reported in patients with AML.5 Molecular investigation of breakpoints in a large number of AML‑associated translocations, insertions, inversions and, occasionally, deletions has resulted in identification of genes involved in leukemogenesis, many of which encode transcription factors whose disruption constitutes a disease‑initiating event. However, in most instances, the presence of a single chromosomal abnormality is not sufficient for the development of an overt AML, which is caused by acquisition of several (on average 13) somatic mutations affecting different pathways. These include, in addition to the aforementioned transcription‑factor fusions, mutations in the NPM1 gene, and mutations in genes belonging to such functional groups as tumor‑suppressors as well as DNA methylation‑related, signaling, chromatin‑modifying, myeloid transcription‑factor, cohesin‑complex, and spliceosome‑complex genes.6
It has been now well established that alterations detectable using cytogenetic methods comprise only a fraction of somatic mutations in patients with AML. However, chromosome aberrations, regardless of whether they are characterized molecularly or not, serve as tumor markers with diagnostic and prognostic significance. A number of more frequent genetic abnormalities, many of which are a result of recurring chromosome abnormalities, are used to define distinct disease entities in the World Health Organization (WHO) Classification of Haematolymphoid Tumours.7 Furthermore, cytogenetic findings at diagnosis have been repeatedly demonstrated to be among the most important, independent prognostic factors for attainment of a complete remission (CR), as well as the disease‑free (DFS) and overall (OS) survival in both younger adults (ie, those younger than 55–60 years) and among older patients (aged 55–60 years and older).3,4,8-18 Also, pretreatment karyotypic data together with molecular genetic findings are used to determine the choice of therapy in these patients.19,20 Consequently, cytogenetic analysis is a mandatory part of the diagnostic work‑up of patients with AML according to the Clinical Practice Guidelines in Oncology of the National Comprehensive Cancer Network19 and the recommendations of an international expert panel working on behalf of the European LeukemiaNet (ELN).20
Disease entities recognized in the World Health Organization classification of acute myeloid leukemia that are defined by gene fusions created by recurrent chromosome aberrations
For the first time, the WHO classification used selected reciprocal translocations and inversions and their molecular counterparts to identify distinct disease entities of AML in 2001. The most recent, 5th edition of this classification7 has increased the role of genetic markers and recognized 11 subgroups within the “AML with defining genetic abnormalities” category, 9 of which are directly associated with chromosome aberrations (Table 1). One of these categories, namely “AML with RBM15::MRTFA,” has been hitherto diagnosed exclusively in children, most of whom were younger than 2 years,5 and will not be described here in more detail.
Data from Khoury et al7
a At least 20% of blasts are required for diagnosis.
b Other recurrent chromosome rearrangements involving 11q23/KMT2A are listed in Table 3.
c Other recurrent chromosome rearrangements involving 11p15/NUP98 are listed in Table 2.
d To diagnose AML, myelodysplasia‑related in patients with 20% of blasts in their bone marrow or blood, the presence of at least 1 cytogenetic or molecular abnormality listed in the second column of the Table and / or history of myelodysplastic or myeloproliferative neoplasm are required.
Abbreviations: AML, acute myeloid leukemia; APL, acute promyelocytic leukemia | |
AML with defining genetic abnormalities | Most frequent cytogenetic findings |
APL with PML::RARA fusion | t(15;17)(q22–24;q12–21) |
AML with RUNX1::RUNX1T1 fusion | t(8;21)(q22;q22.1) |
AML with CBFB::MYH11 fusion | inv(16)(p13.1q22) or t(16;16)(p13.1;q22) |
AML with DEK::NUP214 fusion | t(6;9)(p23;q34.1) |
AML with RBM15::MRTFA fusion | t(1;22)(p13.3;q13.3) |
AML with BCR::ABL1 fusiona | t(9;22)(q34;q11.2) |
AML with KMT2A rearrangement | t(9;11)(p21.3–22;q23.3)b |
AML with MECOM rearrangement | inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2) |
AML with NUP98 rearrangement | t(7;11)(p15;p15)c |
AML with NPM1 mutation | Normal karyotype |
AML with CEBPA mutationa | Normal karyotype |
AML with other defined genetic alterations | Abnormalities creating “rare genetic fusions” |
AML, myelodysplasia‑relatedd | Defining cytogenetic abnormalities |
Complex karyotype (≥3 abnormalities) | |
5q deletion or loss of 5q due to unbalanced translocation | |
Monosomy 7, 7q deletion, or loss of 7q due to unbalanced translocation | |
11q deletion | |
12p deletion or loss of 12p due to unbalanced translocation | |
Monosomy 13 or 13q deletion | |
17p deletion or loss of 17p due to unbalanced translocation | |
Isochromosome 17q | |
idic(X)(q13) | |
Defining somatic mutations in the genes: | |
ASXL1, BCOR, EZH2, SF3B1, SRSF2, STAG2, U2AF1 and / or ZRSR2 | |
Acute promyelocytic leukemia with PML::RARA fusion
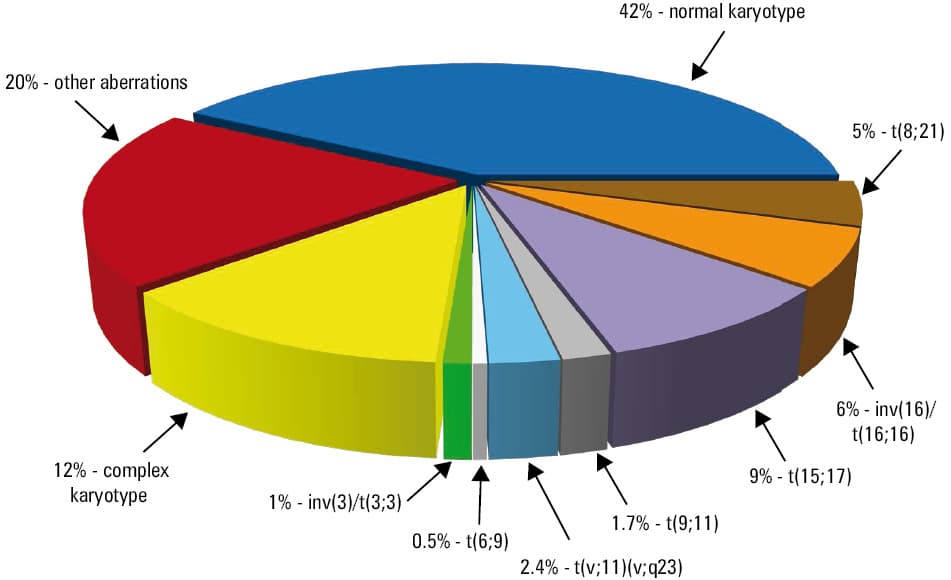
Acute promyelocytic leukemia (APL) is an AML subtype characterized by unique bone marrow (BM) morphology and biological features, which make it a distinctive disease entity that was the first to become curable by therapy targeting causative genetic alterations. Patients with APL constitute 8% to 12% of adults with AML (Figure 1).4,12 These patients invariably harbor a gene fusion between RARA, a gene encoding the retinoic acid receptor α, and a partner gene, which in most cases (99%) is the PML gene. The PML::RARA fusion is generated most often by a reciprocal translocation t(15;17)(q22–24;q12–21) or its infrequent 3- or 4‑way variants that involve, respectively, 1 or 2 additional chromosomes. In a recent study of over 800 patients with PML::RARA fusion,21 96.6% of all cases had a reciprocal t(15;17), including 3.4% of patients with an additional abnormality of either der(17)t(15;17) or der(15)t(15;17). Three- and four‑way variant translocations were detected in 2.2% and 0.1% of cases, respectively, and cryptic PML::RARA fusions with morphologically normal chromosomes 15 and 17 were found in 0.7% of the patients.21 In the latter subgroup, which in an earlier study was estimated to constitute as many as 4% of APL patients,22 the PML::RARA fusion is created by an insertion of a tiny segment containing the RARA gene into the PML locus (or, less frequently, vice versa). Such cryptic rearrangements can be identified using fluorescence in situ hybridization (FISH) or reverse transcription–polymerase chain reaction (RT‑PCR). Importantly, the patients with these hidden alterations do not differ from those harboring standard t(15;17) with regard to clinical features or response to treatment.22
In a small fraction (<1%) of APL patients, RARA is fused with genes other than PML as a result of variant rearrangements that include 7 recurring translocations between chromosome 17 and such partners as chromosomes 3, 4, 5, 11 (twice, with different breakpoints in 11q), 17, and X (Table 2). Determination of the exact gene fusion is vital because this informs the treating physician if the patient can respond to a therapy with all‑trans‑retinoic acid (ATRA) and / or arsenic trioxide (ATO). ATRA has been effective in the patients with the classic PML::RARA fusion and in those with variant fusions between RARA and the BCOR, NPM1, NUMA1, FIP1L1, IRF2BP2, PRKAR1A, and FNDC3B genes.23 In contrast, APL with t(11;17)(q23;q21)/ZBTB16::RARA, t(3;17)(q26;q21)/TBL1XR1::RARA, and t(17;17)(q21;q21)/STAT5B::RARA are resistant to ATRA and associated with an inferior prognosis. Additionally, only APL with PML::RARA, TBL1XR1::RARA, IRF2BP2::RARA, PRKAR1A::RARA, and FNDC3B::RARA has been thus far responsive to treatment with ATO.23
Cytogenetic abnormality | Cases, n | % sole | Gene fusion | Cytogenetic abnormality | Cases, n | % sole | Gene fusion |
Data from Mitelman et al5
a Only the abnormalities for which fusion genes have been identified are included (except for rearrangements involving the MECOM gene).
b Also interpreted as ins(3;3)(q21;q21q26)
c Also interpreted as t(3;5)(q21;q31)
d This inversion is cryptic; there are no individual cases listed in the Mitelman database.5 The numbers of cases listed in the Table are based on data from Gruber et al (doi:10.1016/j.ccr.2012.10.007) and Masetti et al (doi:10.1182/blood‑2012–11–469825).
e In the literature, the breakpoints in t(15;17) have been variously assigned to 15q22 or 15q24, and to 17q11, 17q12, 17q21, or 17q22.
f This translocation is cryptic; there are no individual cases listed in the Mitelman database.5 The numbers of cases listed in the Table are based on data from van Zutven et al (doi:10.1002/gcc.20308), Hollink et al (doi:10.1182/blood‑2011–04–346643), and Gruber et al (doi:10.1016/j.ccr.2012.10.007). | |||||||
Rearrangement involving band 1p36 and the PRDM16 gene | Rearrangement involving band 11q14 and the PICALM gene | ||||||
t(1;3)(p36.3;q21.1) | 56 | 61 | RPN1::PRDM16 | t(10;11)(p12–13;q14–21) | 34 | 35 | MLLT10::PICALM |
Rearrangement involving band 1p13 and the RBM15 gene | Rearrangements involving band 12p13 and the ETV6 gene | ||||||
t(1;22)(p13;q13) | 64 | 63 | RBM15::MRTFA | t(1;12)(q21;p13) | 2 | 50 | ETV6::ARNT |
Rearrangement involving band 2p23 and the ALK gene | t(1;12)(q25;p13) | 2 | 50 | ETV6::ABL2 | |||
inv(2)(p23q13) | 4 | 25 | RANBP2::ALK | t(3;12)(q26;p13) | 45 | 62 | ETV6::MECOM |
Rearrangements involving band 3q26 and the MECOM (EVI1) gene | t(4;12)(q11–12;p13) | 31 | 71 | CHIC2::ETV6 | |||
t(2;3)(p15–21;q26–27) | 21 | 38 | MECOM | t(5;12)(q31;p13) | 5 | 40 | ETV6::ACSL6 |
inv(3)(q21q26.2) | 391 | 35 | RPN1::MECOM GATA2::MECOM | t(7;12)(q36;p13) | 29 | 7 | MNX1::ETV6 |
t(3;3)(q21;q26.2)b | 170 | 41 | RPN1::MECOM GATA2::MECOM | t(8;12)(q12;p13) | 3 | 0 | ETV6::LYN |
t(3;5)(q26–27;q31) | 2 | 50 | MACROH2A1::MECOM | t(10;12)(q24;p13) | 7 | 29 | ETV6::GOT1 |
t(3;8)(q26;q24) | 18 | 56 | MECOM | t(12;13)(p13;q12) | 4 | 50 | ETV6::CDX2 or FLT3::ETV6 |
Rearrangement involving band 5q34 and the NPM1 gene | t(12;22)(p12–13;q11–13) | 29 | 21 | ETV6::MN1 | |||
t(3;5)(q25;q34)c | 78 | 80 | MLF1::NPM1 | Rearrangements involving band 12q13 and the RARG gene | |||
Rearrangement involving band 6q23 and the MYB gene | t(11;12)(p15;q13) | 3 | 100 | NUP98::RARG | |||
t(X;6)(p11;q23) | 5 | 100 | MYB::GATA1 | Rearrangements involving band 16q22 and the CBFB gene | |||
Rearrangements involving band 8p11 and the KAT6A gene | inv(16)(p13.1q22) | 1003 | 68 | MYH11::CBFB | |||
inv(8)(p11q13) | 7 | 86 | KAT6A::NCOA2 | t(16;16)(p13.1;q22) | 52 | 83 | MYH11::CBFB |
t(8;16)(p11;p13) | 148 | 58 | KAT6A::CREBBP | Rearrangements involving band 16q24 and the CBFA2T3 gene | |||
t(8;19)(p11;q13.3) | 6 | 100 | KAT6A::LEUTX | t(1;16)(p31;q24) | 4 | 0 | NFIA::CBFA2T3 |
t(8;22)(p11;q13) | 5 | 60 | KAT6A::EP300 | inv(16)(p13q24)d | 32 | 6 | CBFA2T3::GLIS2 |
Rearrangements involving band 8p11 and the FGFR1 gene | t(16;21)(q24;q22) | 31 | 29 | CBFA2T3::RUNX1 | |||
t(3;8)(q12;p11) | 2 | 0 | TFG::FGFR1 | Rearrangement involving band 17q11 and the TAF15 gene | |||
t(6;8)(q27;p11) | 5 | 60 | CEP43::FGFR1 | t(12;17)(p13;q11–12) | 6 | 100 | TAF15::ZNF384 |
t(8;9)(p11;q33) | 2 | 0 | CNTRL::FGFR1 | Rearrangements involving bands 17q12–21 and the RARA gene | |||
t(8;13)(p11;q12) | 4 | 25 | ZMYM2::FGFR1 | t(1;17)(q42;q21) | 2 | 0 | IRF2BP2::RARA |
t(8;22)(p11;q11) | 4 | 75 | BCR::FGFR1 | t(3;17)(q26;q21) | 3 | 0 | TBL1XR1::RARA or FNDC3B::RARA |
Rearrangement involving band 9p24 and the JAK2 gene | t(4;17)(q12;q21) | 2 | 0 | FIP1L1::RARA | |||
t(8;9)(p22;p24) | 6 | 50 | PCM1::JAK2 | t(5;17)(q35;q12–21) | 4 | 25 | NPM1::RARA |
Rearrangement involving band 9q34 and the NUP214 gene | t(11;17)(q13;q12–21) | 2 | 50 | NUMA1::RARA | |||
t(6;9)(p23;q34) | 116 | 72 | DEK::NUP214 | t(11;17)(q23;q21) | 27 | 78 | ZBTB16::RARA |
Rearrangement involving band 9q34 and the ABL1 gene | t(15;17)(q22–24;q12–21)e | 1380 | 70 | PML::RARA | |||
t(9;22)(q34;q11.2) | 302 | 44 | BCR::ABL1 | t(17;17)(q21;q21) | 3 | 67 | STAT5B::RARA |
Rearrangement involving band 10q22 and the KAT6B gene | t(X;17)(p11.4;q21) | 2 | 0 | BCOR::RARA | |||
t(10;16)(q22;p13) | 4 | 50 | KAT6B::CREBBP | Rearrangements involving band 21q22 and the RUNX1 gene | |||
Rearrangement involving band 10p15 and the ZMYND11 gene | t(1;21)(p36;q22) | 6 | 83 | RUNX1::PRDM16 | |||
t(10;17)(p15;q21) | 12 | 75 | MBTD1::ZMYND11 | t(1;21)(p32;q22) | 6 | 33 | RUNX1::PRPF38A or RUNX1::NDC1 |
Rearrangements involving band 11p15 and the NUP98 gene | t(1;21)(p22;q22) | 2 | 100 | RUNX1::CLCA2 | |||
t(1;11)(q23;p15) | 5 | 40 | NUP98::PRRX1 | t(3;21)(q26;q22) | 70 | 44 | MECOM::RUNX1 |
t(2;11)(q31;p15) | 7 | 86 | NUP98::HOXD11 or NUP98::HOXD13 | t(4;21)(q31;q22) | 2 | 50 | RUNX1::SH3D19 |
t(3;11)(p11;p15) | 2 | 100 | NUP98::POU1F1 | t(5;21)(q23.3–31;q22) | 3 | 67 | RUNX1::ADAMTS19 |
t(3;11)(q12;p15) | 5 | 80 | NUP98::LNP1 | t(7;21)(p22;q22) | 11 | 36 | RUNX1::USP42 |
t(4;11)(q21–23;p15) | 5 | 40 | NUP98::RAP1GDS1 | t(8;21)(p11;q22) | 3 | 33 | RUNX1::TACC1 |
t(5;11)(q35;p15) | 42 | 69 | NUP98::NSD1 or STIM1::NSD1 | t(8;21)(q22;q22) | 1768 | 42 | RUNX1T1::RUNX1 |
t(7;11)(p15;p15) | 79 | 78 | NUP98::HOXA9 or NUP98::HOXA11 or NUP98::HOXA13 | t(8;21)(q23–24;q22) | 3 | 67 | RUNX1::TRPS1 |
t(8;11)(p11;p15) | 5 | 40 | NUP98::NSD3 | t(11;21)(p13–4;q22) | 4 | 50 | RUNX1::KIAA1549L |
t(9;11)(p22;p15) | 7 | 86 | NUP98::PSIP1 | t(11;21)(q13;q22) | 2 | 50 | RUNX1::MACROD1 |
t(9;11)(q34;p15) | 2 | 50 | NUP98::PRRX2 | t(15;21)(q21;q22) | 3 | 33 | RUNX1::TCF12 |
t(10;11)(q23;p15) | 4 | 50 | NUP98::HHEX | t(20;21)(q11;q22) | 2 | 0 | RUNX1::NOL4L or RUNX1::CBFA2T2 |
inv(11)(p15q22) | 17 | 65 | NUP98::DDX10 | Rearrangement involving band 21q22 and the ERG gene | |||
t(11;12)(p15;p13)f | 6 | 17 | NUP98::KDM5A | t(16;21)(p11;q22) | 63 | 67 | FUS::ERG |
t(11;12)(p15;q13) | 14 | 86 | NUP98::HOXC11 or NUP98::HOXC13 | ||||
t(11;17)(p15;p13) | 5 | 100 | NUP98::PHF23 | ||||
t(11;20)(p15;q11–12) | 18 | 78 | NUP98::TOP1 | ||||
The prognosis of patients with APL and t(15;17)/PML::RARA, which historically had been the worst among the subtypes of AML, has become the most favorable when treatment regimens containing ATRA and / or ATO became available, with CR rates of 85% to 100% and a cure rate of up to 97% in recent studies.24 Patients with therapy‑related APL receiving adequate treatment with ATRA‑containing regimens have an outcome comparable to that of patients with de novo disease.25 However, APL patients are prone to coagulopathy and hyperfibrinolysis that may lead to gastrointestinal bleeding and intrapulmonary and / or intracranial hemorrhages, thrombotic events, and early mortality. Therefore, prompt diagnosis and early detection of APL‑associated rearrangements followed by ATRA treatment is vital. The use of RT‑PCR and / or FISH offers potentially quicker results22; however, these assays should not replace but rather supplement conventional cytogenetic analysis. The latter can identify rare (and potentially novel) variant translocations as well as detect secondary cytogenetic changes, which accompany t(15;17) in approximately one‑third of APL patients at diagnosis. These secondary abnormalities most often include +8 or +8q, followed by del(7q)/add(7q) and del(9q).21,26 Complex karyotype [ie, the presence of ≥2 aberrations in addition to t(15;17)] is detected in 8% to 12% of the patients.21,27,28 Although individual secondary abnormalities do not appear to influence patients’ prognosis,22,27,28 a complex karyotype conferred a lower CR rate,27 shorter event‑free survival,28 and shorter OS27 than those in APL patients with a noncomplex karyotype who were treated with regimens containing ATO. This indicates a need for an alternative, more intense treatment when a complex karyotype is detected in an APL patient.
Internal tandem duplications of the FLT3 gene (FLT3-ITD), which confer adverse prognosis in cytogenetically normal AML (CN‑AML), are detected in 20% to approximately 40% of APL patients and have been associated with variant microgranular (or hypogranular) BM morphology and higher white blood cell (WBC) counts at diagnosis.29 Although FLT3-ITD has been associated with higher frequency of induction death, CR rates have not been affected by its presence and the adverse impact of FLT3-ITD on survival, shown in some but not all studies, remains debatable.29 Moreover, treatment with ATO appears to overcome the potentially negative influence of FLT3-ITD on patients’ survival.24,27
Acute myeloid leukemia with RUNX1::RUNX1T1 fusion
The presence of t(8;21)/RUNX1::RUNX1T1 denotes one of the 2 AML subtypes that constitute the so‑called core‑binding factor AML (CBF‑AML), the second being AML with inv(16)(p13.1q22). Both chromosome aberrations result in rearrangements of genes encoding the α and β subunits of the CBF complex, respectively, the RUNX1 gene, located at 21q22.1 and disrupted by t(8;21) and the CBFB gene, located at 16q22 and disrupted by inv(16)/t(16;16). The CBF complex is a heterodimeric transcription factor regulating transcription of genes encoding proteins involved in hematopoietic differentiation. A protein encoded by a chimeric gene RUNX1::RUNX1T1 created by t(8;21) inhibits the wild‑type RUNX1 in a dominant‑negative fashion and leads to impairment of normal hematopoiesis and predisposition to develop leukemia.30
Translocation t(8;21) or its infrequent variants [eg, ins(8;21)(q22;q22q22), ins(21;8)(q22;q21q22), t(8;10;21)(q22;q26;q22), t(8;20;21)(q22;p11.2;q22), etc] are found in 5% to 8% of adults with AML (Figure 1).12,31 Only roughly 30% of the patients harbor t(8;21) as the only chromosome abnormality. The most frequent secondary aberration is –Y, detected in about 60% of male patients, followed by –X, found in 33% to 40% of women with t(8;21), and by del(9q) (17%), +8 (5%–7%), and +4 (4%).32,33 Evidently, additional genetic alterations cooperating with RUNX1::RUNX1T1 are required because the presence of this gene fusion alone has not been sufficient to induce leukemia.34 Potential genetic rearrangements that cooperate with t(8;21)/RUNX1::RUNX1T1 include KIT mutations (found in ca 25% of patients), NRAS and KRAS mutations (10%–20%), FLT3-ITD (7%), and point mutations in the tyrosine kinase domain of FLT3 (FLT3-TKD; 4%).34-36
Treatment outcome of patients with t(8;21) receiving postremission therapy with repetitive cycles of high‑dose cytarabine (HiDAC) is relatively favorable,32-34,37 albeit OS and survival after the first relapse of patients with t(8;21) are shorter than those of patients with inv(16).33 Since secondary aberrations are present in most patients, their prognostic significance has been of major interest and was assessed by many studies with conflicting results. Loss of the Y chromosome was reported as having adverse,32,38 favorable,39 or no33,40 significant impact on the outcome of men with t(8;21). Likewise, loss of the X chromosome was recently associated with better outcome in a large cohort of female patients with t(8;21) in China,41 but this has not been observed in European or American studies.32,33,39 Deletion of 9q was found to confer favorable prognosis in 2 studies,33,40 but this effect was limited to non‑White patients in one of them.33 On the other hand, worse outcome has been repeatedly associated with KIT mutations.34,35 Moreover, high cumulative incidence of relapse (CIR), but not shorter OS, was associated with higher (≥25%) relative KIT mutant levels only.42 Likewise, high levels of FLT3-ITD resulted in shorter OS, but not higher CIR, in younger adults with t(8;21).42 These results should be further corroborated.
Acute myeloid leukemia with CBFB::MYH11 fusion
The second subset of CBF‑AML is defined by the presence of CBFB::MYH11 gene fusion created by either inv(16)(p13.1q22) or, less commonly, t(16;16)(p13.1;q22). These abnormalities are seen in 5% to 6% of adults with AML (Figure 1), and are closely associated with myelomonocytic BM morphology and abnormal eosinophils, which are pathognomonic for this type of AML.12 The CBFB::MYH11 fusion gene encodes a chimeric protein that retains the ability to interact with RUNX1 and block CBF‑dependent transcription. Due to the marked variability of genomic breakpoints within the CBFB and MYH11 genes, over 10 fusion transcript variants of different sizes have been reported.43 The most frequent type A fusion is detected in 85%, and type D and type E fusions are each found in 5% to 10% of the patients. For still unclear reasons, the patients harboring type A fusions differ from those with non–type A fusions with regard to the occurrence of additional genetic alterations. In addition to higher WBC counts, the patients with type A fusions had less often secondary +8 or +21, but only they carried +22 and the KIT mutations, which were not detected in patients with non–type A fusions. Mutations in the KIT gene bestowed adverse prognosis among type A fusion–positive patients, whereas treatment outcomes of the type A and non–type A patients with wild‑type KIT were not significantly different.43
Prognosis of patients with inv(16)/t(16;16) is relatively favorable, especially if they are treated with 3 to 4 cycles of HiDAC postremission.44 Recent studies demonstrated that clinical outcome of CBF‑AML patients with inv(16)/t(16;16) and those with t(8;21) can be further improved by the addition of dasatinib, a multi‑kinase inhibitor, to intensive chemotherapy.45,46 Prognosis for therapy‑related CBF‑AML with inv(16)/t(16;16) or t(8;21) seems to be worse than for de novo CBF‑AML, but still better than for therapy‑related non–CBF‑AML.25
The presence of secondary +22 has been repeatedly found to reduce the patients’ risk of relapse,32-34 lengthen their OS duration13,34 and, in patients who also carry FLT3-ITD, extend relapse‑free survival.47 Among molecular markers, mutations in KIT34,35 and FLT3, mainly FLT3-TKD,34 adversely affect OS. Although NRAS and KRAS mutations are frequent (seen in >50% of patients), they do not influence prognosis.34,42,48 However, RAS mutations seem to sensitize AML blasts to postremission therapy with HiDAC.49
Acute myeloid leukemia with DEK::NUP214 fusion The t(6;9)(p23;q34.1), which generates the DEK::NUP214 fusion, is fairly rare, being detected in 0.5% to 0.7% of adults with AML (Figure 1). In approximately 90% of cases, it is the sole chromosome abnormality,5,50 indicating that t(6;9)/DEK::NUP214 represents the main driver event in leukemogenesis. Although secondary chromosome aberrations are rare, patients with t(6;9) or its infrequent 3‑way variants harbor FLT3-ITD more often than individuals with any other AML subtype, with 73% to 85% of t(6;9)-positive adults acquiring this mutation.50,51
The clinical outcome of adult patients receiving chemotherapy is very poor irrespective of the presence or absence of FLT3-ITD.50 In contrast, allogeneic hematopoietic stem cell transplantation (alloHSCT) performed in patients in CR has been reported to substantially improve their prognosis.51,52
Acute myeloid leukemia with KMT2A rearrangement Band 11q23.3 is a locus of the KMT2A gene (formerly MLL), which is fused with the highest number of partner genes in human leukemia as a result of balanced chromosome abnormalities occurring both in AML and acute lymphoblastic leukemia. In AML, at least 77 different fusion partners of 11q23/KMT2A have been reported, some in single patients.3 It is likely that further rearrangements involving 11q23/KMT2A and novel partner genes will be discovered using targeted next‑generation sequencing (NGS) in addition to the conventional cytogenetic, FISH, and long‑distance inverse (LDI)-PCR assays.54 Table 3 lists 43 reciprocal translocations, insertions, inversions, and deletions involving 11q23/KMT2A, together with gene fusions they created. Only recurrent abnormalities found in at least 2 cases of AML are displayed.
Cytogenetic abnormality | Cases, n | % sole | Gene fusion | Cytogenetic abnormality | Cases, n | % sole | Gene fusion |
Data from Mitelman et al5
a Only the abnormalities for which fusion genes have been identified are included.
b The numbers of cases listed in the Table include data from Kourlas et al (doi:10.1073/pnas.040569197) and Shih et al (doi:10.1038/sj.leu.2404024) | |||||||
t(1;11)(p32;q23) | 16 | 56 | KMT2A::EPS15 | t(11;11)(q13;q23) | 3 | 67 | KMT2A::C2CD3 or KMT2A::ARHGEF17 |
t(1;11)(q21;q23) | 31 | 71 | KMT2A::MLLT11 | inv(11)(q21q23) | 7 | 43 | KMT2A::MAML2 |
t(2;11)(q37;q23) | 8 | 63 | KMT2A::SEPTIN2 | t(11;12)(q23;q13) | 2 | 100 | KMT2A::SARNP |
t(3;11)(p21;q23) | 2 | 50 | KMT2A::NCKIPSD | t(11;14)(q23;q23–24) | 3 | 100 | KMT2A::GPHN |
t(4;11)(q21;q23) | 39 | 49 | KMT2A::AFF1 | t(11;14)(q23;q32) | 3 | 100 | KMT2A::CEP170B |
t(5;11)(q31;q23) | 6 | 100 | KMT2A::ARHGAP26 | t(11;15)(q23;q14–15) | 11 | 18 | KMT2A::KNL1 or KMT2A::ZFYVE19 |
t(6;11)(q15;q23) | 3 | 67 | KMT2A::CASP8AP2 | t(11;16)(q23;p13) | 14 | 50 | KMT2A::CREBBP or KMT2A::MYH11 |
t(6;11)(q21;q23) | 6 | 33 | KMT2A::FOXO3 | t(11;16)(q23;q24) | 3 | 67 | KMT2A::USP10 |
t(6;11)(q25;q23) | 2 | 100 | KMT2A::SNX9 | t(11;17)(q23;p13) | 2 | 100 | KMT2A::GAS7 |
t(6;11)(q27;q23) | 118 | 81 | KMT2A::AFDN | t(11;17)(q23;q12–21) | 67 | 81 | KMT2A::MLLT6 or KMT2A::RARA or KMT2A::ACACA |
t(9;11)(p21–22;q23) | 462 | 67 | KMT2A::MLLT3 | t(11;17)(q23;q23) | 4 | 50 | KMT2A::CLTC |
t(9;11)(q33–34;q23) | 2 | 100 | KMT2A::DAB2IP | t(11;17)(q23;q25) | 45 | 67 | KMT2A::SEPTIN9 |
ins(10;11)(p11–13;q23q13–25) | 33 | 45 | KMT2A::MLLT10 | t(11;18)(q23;q21) | 3 | 33 | KMT2A::ME2 |
ins(11;10)(q23;p12–13p11–15) | 8 | 75 | KMT2A::MLLT10 | t(11;19)(q23;p13.1) | 69 | 85 | KMT2A::ELL |
t(10;11)(p12;q23) | 3 | 100 | KMT2A::ABI1 or KMT2A::NEBL | t(11;19)(q23;p13.3) | 47 | 41 | KMT2A::MLLT1 or KMT2A::VAV1 |
t(10;11)(p11–13;q13–23) | 60 | 48 | KMT2A::MLLT10 | t(11;19)(q23;p13.2–13.3) | 4 | 25 | KMT2A::MYO1F |
t(10;11)(q21–22;q23) | 9 | 67 | KMT2A::TET1 | t(11;22)(q23;q11) | 13 | 62 | KMT2A::SEPTIN5 |
del(11)(q23q23)b | 5 | 20 | KMT2A::ARHGEF12 | t(11;22)(q23;q13) | 6 | 67 | KMT2A::EP300 |
del(11)(q23q24) | 4 | 50 | KMT2A::DCPS or KMT2A::TIRAP | t(X;11)(q13;q23) | 3 | 100 | KMT2A::FOXO4 |
inv(11)(p15q23) | 13 | 69 | KMT2A::NRIP3 or NUP98::KMT2A | t(X;11)(q24;q23) | 4 | 100 | KMT2A::SEPTIN6 |
t(11;11)(p15;q23) | 3 | 100 | KMT2A::NRIP3 or KMT2A::AP2A2 | ins(11;X)(q23;q28q12) | 3 | 67 | KMT2A::FLNA |
inv(11)(q13q23) | 3 | 0 | KMT2A::C2CD3 | ||||
Translocation t(9;11) is the most frequent among 11q23/KMT2A rearrangements, being detected in approximately 2% of adults with AML (Figure 1). This translocation fuses KMT2A, the gene encoding a DNA‑binding protein methylating histone H3 lysine 4 (H3K4) and positively regulating the expression of multiple genes including the HOX genes, with MLLT3, a gene encoding a nuclear protein containing serine‑rich and proline‑rich regions, which appear to be important for leukemogenesis.55 The t(9;11) is the sole chromosome change in nearly two‑thirds of patients; around 20% of cases have +8, and less frequent are secondary +19 and +21.5
Several studies reported that the patients with t(9;11) had better treatment outcome than those with other rearrangements involving 11q23/KMT2A,11,13,56-59 which placed the patients with t(9;11) in the intermediate‑risk category in both prognostic categorizations based solely on cytogenetic data11,13 and in classifications combining cytogenetics with selected molecular genetic findings, such as the 201060 and 202220 ELN classifications. However, a more favorable prognosis of the patients with t(9;11) was observed in studies analyzing exclusively13,57 or mostly11,56,58 adults younger than 60 years. When the analysis was restricted to patients aged 60 years or older, the outcome of the t(9;11)-positive patients was very poor and comparable to that of individuals with other 11q23/KMT2A alterations,59,61 whose prognosis is invariably adverse regardless of the age group.11-13,56,59,61 The reasons for such age difference are presently unknown.
Acute myeloid leukemia with MECOM rearrangement
The MECOM rearrangements are caused most often by inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2). Both inv(3) and the less frequent t(3;3) cause relocation of a distal enhancer of the GATA2 gene that leads to simultaneous deregulated expression of the MECOM gene (formerly known as EVI1) and haploinsufficiency of GATA2. The inv(3)/t(3;3) are detected in approximately 1% of patients with AML (Figure 1), both de novo and evolving from preceding myelodysplastic syndrome, and are associated with multilineage dysplasia, abnormal megakaryopoiesis with micromegakaryocytes in the BM, normal or increased platelet counts, and higher WBC counts at diagnosis.62 The majority of patients have secondary abnormalities, with −7 being found in approximately 50% of cases.5 Since inv(3) is a subtle rearrangement, which can be illustrated by the fact that it was occasionally missed by a cytogenetic laboratory to be later recognized on central karyotype review performed by the Cancer and Leukemia Group B / Alliance for Clinical Trial in Oncology (CALGB/Alliance),63 detection of an apparently sole −7 should always prompt cytogeneticists to closely inspect chromosomes 3 in such patients.
The treatment outcome of the patients with inv(3)/t(3;3) is invariably dismal regardless of the presence or absence of −7 in most studies,11,13 although Lugthart et al62 reported that patients with −7 fared even worse than those without −7. To date, alloHSCT, especially if performed in CR, appears to be the only option to improve the very poor prognosis of patients with inv(3)/t(3;3).64
Acute myeloid leukemia with NUP98 rearrangement
NUP98 participates in over 30 gene fusions with different partner genes that are generated by reciprocal translocations or inversions, the recurrent of which are listed in Table 2. Because NUP98 is located near the terminus of 11p, at 11p15.4, some 11p15.4/NUP98 alterations may be undetectable by conventional karyotyping.7 AML with NUP98 rearrangement is usually characterized by deregulation of the HOXA gene cluster and decreased terminal differentiation of hematopoietic cells.65 The majority of specific abnormalities involving 11p15.4/NUP98 are quite rare and occur considerably more often in children than adults.66 Although these abnormalities bestow poor prognosis in children, their prognostic significance is yet to be conclusively established in large cohorts of adult patients.
Acute myeloid leukemia, myelodysplasia‑related
This is another WHO category of AML that employs cytogenetic information to classify patients lacking any of the abnormalities described above.7 To be assigned to this AML subtype, it is sufficient for the patient whose BM or blood contains at least 20% of blasts to harbor a complex karyotype with 3 or more chromosome aberrations, and / or one of 8 unbalanced abnormalities or a somatic mutation in one of the following genes: ASXL1, BCOR, EZH2, SF3B1, SRSF2, STAG2, U2AF1, and ZRSR2 (Table 1). Most of the unbalanced aberrations, except for idic(X)(q13) or del(11q), occur frequently as part of a complex karyotype and their presence negatively affects patient prognosis.67
Patients who do not carry any of the aforementioned defining genetic alterations or whose pretreatment cytogenetic investigation fails are categorized into AML subtypes defined by differentiation.7 Notably, unsuccessful cytogenetic assays are rare, occurring in 2% to 6% of patients in large studies.68,69
Complex karyotype and its clinical significance in acute myeloid leukemia
Most studies define complex karyotype as the one encompassing at least 3 chromosome aberrations,9,11,15-17,70 although definitions of 5 or more unrelated cytogenetic abnormalities14,16 and 4 or more abnormalities (excluding the chromosome changes that confer a favorable or adverse prognosis)13 have also been used. The complex karyotype subset usually does not include patients with t(15;17), t(8;21), inv(16)/t(16;16),11,15 t(9;11),11,16 or other balanced rearrangements affecting band 11q23.15,16 Recently, the Francophone Group of Hematological Cytogenetics suggested71 that to standardize the cytogenetic practice, a complex karyotype with 3 chromosome abnormalities should be defined as a low, one with 4 aberrations as an intermediate, and one with 5 or more abnormalities as a highly complex karyotype. The most recent definition proposed by the ELN denotes a complex karyotype as containing at least 3 abnormalities in the absence of “other class‑defining recurring genetic abnormalities” and does not consider karyotypes with 3 or more trisomies in the absence of structural abnormalities as complex.20
Patients with AML and a complex karyotype defined as at least 3 chromosome aberrations constitute 10% to 12%, and those with 5 or more aberrations 8% to 9% of all AML patients.9,11,14 The incidence of AML with a complex karyotype increases with advancing age of the patients and is more than twice as high in patients older than 60 years (17%–19%) than in younger adults (6%–8%).70 Although the lowest number of aberrations in a complex karyotype is 3, in most patients more aberrations are present, with the median numbers being 6,72 8,73 and 10,74 respectively, in 3 studies analyzing complex karyotypes using multiplex FISH or spectral karyotyping. These molecular cytogenetic techniques were instrumental in a more precise delineation of chromosome abnormalities in complex karyotypes, including marker chromosomes, and revealed the nonrandom nature of the occurrence of particular structural and numerical abnormalities in complex karyotypes. Most recurring chromosome alterations are unbalanced (monosomies, deletions, and unbalanced translocations) and result in a loss of chromosome segments mainly from, in decreasing order, chromosome arms 5q, 17p, 7q, 18q, 16q, 17q, 12p, 20q, 18p, and 3p.70,72-75 The loss of the 17p13 locus has been correlated with a loss of and mutations in the TP53 gene, which cause a loss of the p53 protein function. This results in increased cell survival that contributes to pronounced genomic instability and poor outcome of patients with a complex karyotype.76,77 Gains of chromosomal material occur less often and are frequently hidden in unbalanced translocations and marker chromosomes. These gains involve most often 8q, 11q, 21q, 22q, 1p, 9p, and 13q,70,72-75 and their further analysis has led to identification of genes residing in amplified regions such as MYC in 8q, DDX6, ETS1, FLI1, and KMT2A in 11q, ERG, ETS2, and APP in 21q, and CDX2 in 13q12.70,75,78-80
Among unbalanced rearrangements, the most frequent are those leading to a loss of the 5q material that are found in approximately 80% of the patients, followed by losses of 7q and 17p, each detected in circa 50% of the patients. These abnormalities often coexist in the same individual, resulting in around 85% of all patients with complex karyotypes carrying at least one of them.70 Recently, we designated such complex karyotypes with the loss of 5q, 7q, and / or 17p as typical, and complex karyotypes with at least 3 abnormalities that do not include 5q, 7q, and / or 17p loss as atypical.81 An example of a typical complex karyotype with abnormalities that caused the loss of segments from 5q and 17p, in addition to other chromosome rearrangements, is shown in Figure 2. Our comparison of pretreatment characteristics and outcomes of patients with these 2 complex karyotype types revealed that AMLs with typical and atypical complex karyotypes constitute different disease subtypes. In comparison with patients with a typical complex karyotype, those with an atypical one were younger, had higher WBC counts and percentages of blood and BM blasts, harbored TP53 mutations less frequently (10% vs 67%) and more frequently carried FLT3-TKD and PHF6, MED12, and NPM1 mutations. Importantly, they had higher CR rates and longer OS.81 Similarly, in another study,82 the patients with a hyperdiploid complex karyotype (ie, with 49–65 chromosomes) that included exclusively numerical abnormalities that would be recognized as an atypical complex karyotype, had a relatively long OS comparable to the OS of the patients classified in the intermediate cytogenetic risk group as defined by the United Kingdom Medical Research Council trials.13
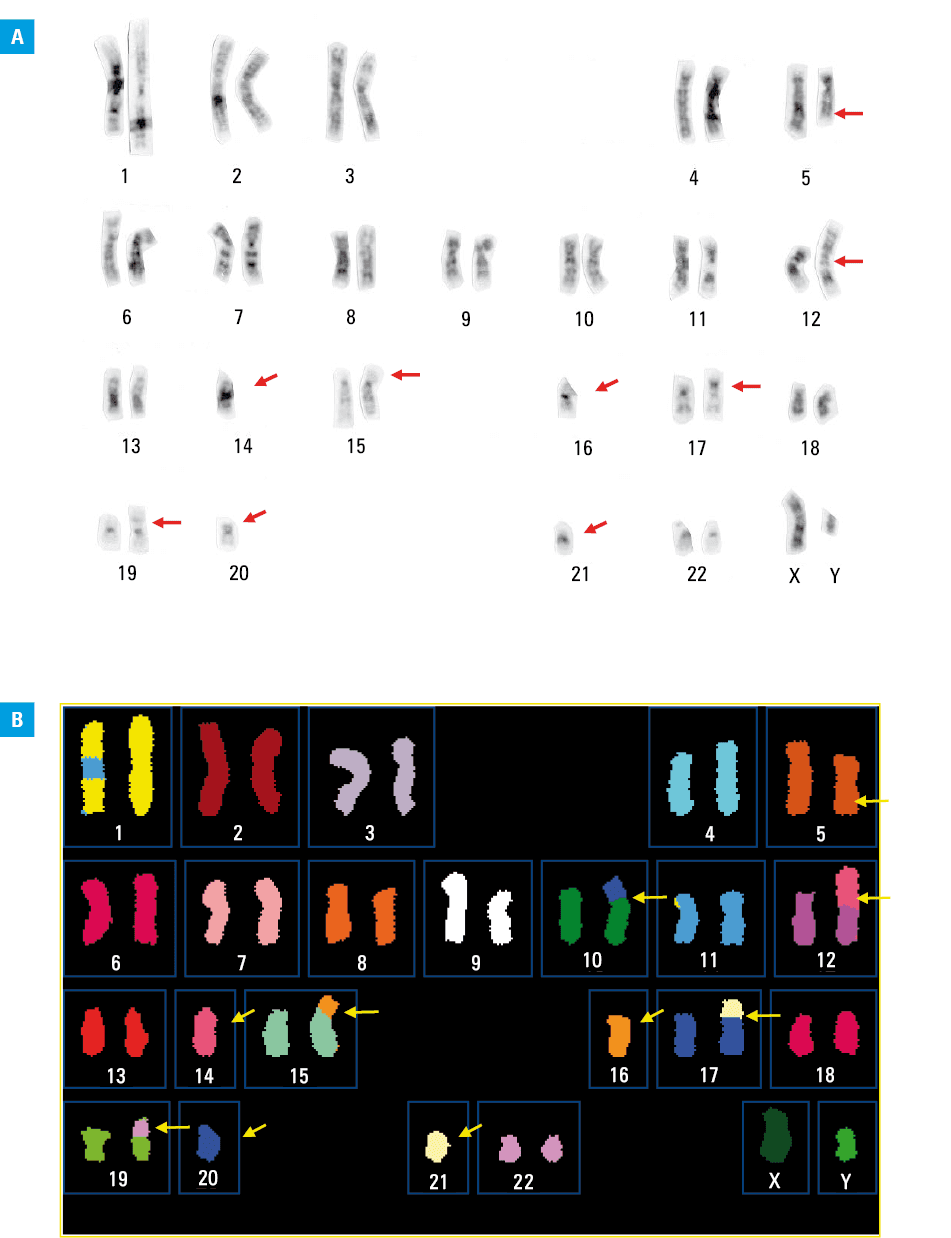
Cytogenetically normal acute myeloid leukemia
The largest cytogenetic subset of adult AML consists of 40% to 45% of patients with a normal karyotype (Figure 1).3,4,12 The proportion of patients with CN‑AML recognized by standard G‑banding analysis is likely overestimated because sometimes subtle aberrations such as inv(16), inv(3) or t(11;19)(q23;p13.1) might be overlooked in suboptimal‑quality preparations. Thus, to eliminate this possibility, CALGB / Alliance has been conducting central karyotype reviews for almost 40 years.63 Moreover, occasionally, CN‑AML patients harbor the abovementioned cryptic insertions producing AML‑associated gene fusions, such as PML::RARA or CBFB::MYH11, albeit patients with these alterations constitute only a fraction of all CN‑AML cases.21,22,31 However, there are also recurring cryptic rearrangements involving terminal bands with similar morphology that are, by definition, undetectable on routine cytogenetic investigation (but detectable by FISH or NGS‑based methods), such as a prognostically adverse t(5;11)(q35;p15)/NUP98::NSD1, detected in 2% of adults with CN‑AML, or a rare t(11;12)(p15;p13)/NUP98::KDM5A (Table 2). Moreover, the genome‑wide copy number analysis with paired normal and leukemic DNA using single nucleotide polymorphism (SNP) arrays and ultra‑dense array comparative genomic hybridization (CGH) platform revealed acquired copy number alterations (CNAs) in 24% and uniparental disomy (UPD) segments in 14% of the study patients with CN‑AML.83 UPDs were found with even higher frequency (26%) in a subsequent study,84 which demonstrated that the most common were UPDs affecting 13q with the locus of the FLT3 gene (found in 7.5% of the patients), 6p (2.8%), and 11p (2.8%), and that their presence was associated with treatment outcome in patients with CN‑AML aged under 60 years. Specifically, UPD of 11p conferred longer OS and UPD of 13q conferred shorter DFS and OS.84 CNAs in AML can also be detected by the relatively fast and cost‑effective multiplex ligation‑dependent probe amplification (MLPA) assay, as shown recently.85 Still, karyotypes of a relatively large proportion of AML patients are truly normal (within resolution of metaphase karyotyping), which, of course, does not preclude the existence of molecular genetic alterations.
In all major cytogenetic risk classifications, patients with CN‑AML were classified as having intermediate risk, because their CR rates, DFS, and OS were inferior to those of adequately treated patients with favorable t(15;17), inv(16), and t(8;21), but better than the outcome of patients with adverse chromosome aberrations.9-13 However, CN‑AML is very heterogeneous at the molecular level and consists of molecular genetic subsets with greatly differing prognoses.86,87 In most instances, NPM1 mutations as well as CEBPA bZIP in‑frame mutations and high expression of miR‑181a bestow good prognosis, whereas poor treatment outcome is associated with MLL-PTD and FLT3-ITD, as well as mutations in the ASXL1, BCOR, DNMT3A (both R882 and non‑R882 mutations), IDH1, IDH2, RUNX1, TET2, and WT1 genes and high expression of BAALC, DNMT3B, ERG, MN1, SPARC, miR‑155, and miR‑3151.20,69,86,87 Because patients with CN‑AML may harbor multiple mutations and / or gene expression changes with prognostic import, various combinations of molecular alterations might modify the prognostic impact of individual abnormalities. This makes research aimed at determination of how combinations of molecular genetic and cytogenetic alterations affect prognosis of both CN‑AML and cytogenetically abnormal patients necessary. Additionally, it is important to underscore the fact that prognostic factors in AML depend on the types of induction and postremission therapy. A prognostically adverse genetic alteration may portend a more favorable outcome when another treatment is given. This includes the use of compounds specifically targeting specific alterations, such as midostaurin and gilteritinib in patients with FLT3-ITD or FLT3-TKD, ivosidenib in IDH1-mutated, or enasidenib in IDH2-mutated patients.88
European LeukemiaNet genetic‑risk classifications combining cytogenetic abnormalities with selected gene mutations
In 2010, the ELN introduced a genetic risk classification, which divided patients with AML into 4 genetic groups: favorable, intermediate I, intermediate II, and adverse, based on a combination of cytogenetic findings and 3 molecular genetic markers, namely, FLT3-ITD as well as NPM1 and CEBPA mutations, which were assessed only in patients with CN‑AML.60 Thereafter, 2 large studies, each analyzing over 1500 patients with AML,61,89 demonstrated that the 2010 ELN classification60 was able to reliably separate the favorable and adverse genetic groups from each other and from both intermediate I and intermediate II groups for such outcome end points as CR rates,61 DFS,61 relapse probability,89 and OS.61,89 Multivariable analyses showed that the association of the 2010 ELN genetic groups with clinical outcome was independent from other established prognostic factors.61
In 2017, the ELN classification90 was modified by, among others, merging 2 intermediate groups into 1, removing the requirement for mutation analysis to be performed only in CN‑AML, substituting biallelic CEBPA mutations for any CEBPA mutation, requiring determination of low and high allelic ratios for FLT3-ITD, and adding ASXL1, RUNX1, and TP53 mutations as further adverse markers to be evaluated. Subsequently, several studies undertook validation of this classification in large series of patients with AML and suggested its refinements.91-94 The recently prepublished 2022 ELN classification20 further increased the role of gene mutations in prognostic stratification of AML. Details of the 2022 ELN classification are provided in Table 4. It remains to be established whether the 2022 changes have improved the usefulness of the ELN genetic risk classification.
Risk category | Genetic abnormality |
Data from Döhner et al20
a Co‑existing mutations in the KIT and / or FLT3 genes do not change the risk classification.
b The presence of the NPM1 mutation in patients with adverse‑risk chromosome abnormalities does not change the patients’ categorization in adverse‑risk group.
c Only in‑frame CEBPA mutations in the basic leucine zipper (bZIP) region are associated with good outcome.
d The presence of t(9;11) outweighs the presence of rare co‑existing gene mutations denoting adverse risk.
e Complex karyotype is defined as ≥3 unrelated chromosome abnormalities, with the exception of karyotypes containing any of the class‑defining recurring genetic abnormalities20 or karyotypes with 3 or more trisomies in the absence of structural abnormalities.
f Monosomal karyotype is defined as a karyotype with ≥2 autosomal monosomies or 1 autosomal monosomy and ≥1 structural chromosome abnormality other than t(8;21) or inv(16) or t(16;16).
g For now, these gene mutations should not be used as adverse prognostic markers if they co‑exist with abnormalities denoting favorable risk.
h At a variant allele fraction of ≥10%, regardless of whether the TP53 mutation is mono- or biallelic. | |
Favorable | t(8;21)(q22;q22.1)/RUNX1::RUNX1T1a |
inv(16)(p13.1q22) or t(16;16)(p13.1;q22)/CBFB::MYH11a | |
Mutated NPM1b without FLT3-ITD | |
bZIP in‑frame mutated CEBPAc | |
Intermediate | Mutated NPM1b with FLT3-ITD |
Wild‑type NPM1 with FLT3-ITD | |
t(9;11)(p21.3;q23.3)/MLLT3::KMT2Ad | |
Cytogenetic and / or molecular abnormalities not classified as favorable or adverse | |
Adverse | t(6;9)(p23;q34.1)/DEK::NUP214 |
t(v;11)(v;q23.3); KMT2A-rearranged | |
t(9;22)(q34.1;q11.2)/BCR::ABL1 | |
t(8;16)(p11;p13)/KAT6A::CREBBP | |
inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2)/GATA2, MECOM (EVI1) | |
t(3;v)(q26.2;v)/MECOM (EVI1)-rearranged | |
–5 or del(5q); –7; –17/abn(17p) | |
Complex karyotype,e monosomal karyotypef | |
Mutated ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1, or ZRSR2g | |
Mutated TP53h | |
Summary and future directions
Cytogenetic research of AML identified many recurrent chromosomal aberrations with a diagnostic and prognostic value. Cytogenetic analysis of pretreatment samples is now obligatory in the diagnostic workup of patients with AML. Karyotypic findings are being used together with the mutational status of several leukemia‑associated genes in such genetic risk classifications as the ones published by the ELN20,60,90 to guide the selection of the most effective treatment. However, the prognostic significance of some less common chromosome abnormalities, many of which are categorized in the intermediate‑risk category, has not yet been ascertained, thus supporting the need for large collaborative studies. Additionally, continuing prospective studies is necessary to correlate cytogenetic and molecular genetic alterations with clinical outcomes of patients treated with novel and / or experimental therapies targeting specific genetic rearrangements.95
This makes correct detection of acquired genetic alterations of utmost importance. Recent years have seen an explosion of studies using high‑throughput NGS technologies, including whole‑genome sequencing (WGS), to analyze AML genomes and characterize complex interactions of genetic variations in leukemogenesis.6,66,96-99 Duncavage et al99 demonstrated the clinical utility of WGS for the evaluation of patients with AML and suggested WGS to be an alternative to cytogenetic analysis of myeloid malignancies. The advantages and limitations of this and other novel technologies100 in hematologic malignant disorders have been recently reviewed in depth by Akkari et al.68 The authors discussed clinical, logistic, technical, and financial implications and concluded that although clinical usefulness of WGS is undisputable, the implementation of WGS technology by clinical laboratories worldwide is currently unlikely. At least in the near future, standard chromosome banding analysis, FISH, and SNP arrays will remain the gold standard of cytogenetic testing in AML and other hematologic neoplasms. It is hoped that further development of emerging technologies may soon enable the use of tools allowing simultaneous delineation of transcriptomic, epigenomic, and proteomic characteristics of leukemic blasts that have clinical significance in AML.
- de la Chapelle A, Schröder J, Vuopio P. 8‑trisomy in the bone marrow. Report of two cases. Clin Genet. 1972; 3: 470‑476. | Crossref
- Rowley JD. Identification of a translocation with quinacrine fluorescence in a patient with acute leukemia. Ann Genet. 1973; 16: 109‑112.
- Mrózek K, Heinonen K, de la Chapelle A, Bloomfield CD. Clinical significance of cytogenetics in acute myeloid leukemia. Semin Oncol. 1997; 24: 17‑31.
- Grimwade D, Mrózek K. Diagnostic and prognostic value of cytogenetics in acute myeloid leukemia. Hematol Oncol Clin North Am. 2011; 25: 1135‑1161, vii. | Crossref
- Mitelman F, Johansson B, Mertens F, eds. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. https://mitelmandatabase.isb‑cgc.org. Accessed July 9, 2022.
ARTICLE INFORMATION