Neuropsychiatric manifestations and their attribution to systemic lupus erythematosus: a retrospective single-center study in a Polish population
Key words: Italian model, neuropsychiatric manifestation attribution, systemic lupus erythematosus



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Neuropsychiatric manifestations and their attribution to systemic lupus erythematosus: a retrospective single-center study in a Polish population
Introduction: Neuropsychiatric (NP) manifestations occur in patients with systemic lupus erythematosus (SLE), and it is challenging to distinguish these manifestations from other neuropsychiatric conditions.
Objectives: We aimed to assess the prevalence of primary neuropsychiatric SLE (NPSLE) in a Polish cohort of SLE patients.
Patients and methods: This retrospective, cross‑sectional study evaluated 164 patients with SLE. NP manifestations were attributed to SLE using the Italian model. Demographic and clinical data, including disease activity (measured by the Systemic Lupus Erythematosus Disease Activity Index version 2000 [SLEDAI‑2K] and the Physician Global Assessment) and organ damage (measured by the Systemic Lupus International Collaborating Clinics / American College of Rheumatology Damage Index), were obtained in patients with and without NP manifestations attributed to SLE.
Results: The final analysis set included 143 patients, 34 of whom (23.8%) had NP manifestations attributed to SLE. The age of the patients with NPSLE and the age of disease onset were significantly lower in comparison with those without NP symptoms attributed to SLE (median [interquartile range], 38 [29–45] vs 45 [32–55] years; P = 0.009, and 35 [24–38] vs 40 [25–48] years; P = 0.03, respectively). The disease activity and proportion of patients with active disease (SLEDAI‑2K ≥6) was significantly higher in the NPSLE patients than in those without NP symptoms attributed to SLE (P <0.005; 100% vs 85.3%; P = 0.01, respectively). NP manifestations in the central nervous system were the most frequent (91.5%). In the patients with NPSLE, cerebrovascular disease, seizures, cognitive dysfunction, psychosis, and cranial neuropathy occurred most often.
Conclusions: NP manifestations occurred mainly in young patients with high disease activity. Cerebrovascular disease, seizures, psychosis, cognitive dysfunction, and cranial neuropathy were the most frequent manifestations of NPSLE.
What's new?
In this retrospective, cross‑sectional study, we assessed the prevalence of primary neuropsychiatric (NP) systemic lupus erythematosus (SLE) using the Italian attribution model in a Polish cohort of patients with SLE. To the best of our knowledge, there are no published studies on the attribution of NP manifestations to primary NPSLE that would also include a detailed characterization of the disease activity and symptoms in Polish patients. Our study demonstrated that primary NP manifestations occurred mainly in young patients with high disease activity (measured by the Systemic Lupus Erythematosus Disease Activity Index version 2000 [SLEDAI‑2K], SLEDAI‑2K without NP manifestations, clinical SLEDAI‑2K, and the Physician Global Assessment). The most frequent manifestations of NPSLE were cerebrovascular disease, seizures, psychosis, cognitive dysfunction, and cranial neuropathy.
Introduction
Neuropsychiatric (NP) manifestations occur in patients with systemic lupus erythematosus (SLE) and may or may not result from the autoimmune processes developing throughout the SLE course. Correct attribution of NP manifestations is challenging, and thus, the diagnosis of primary NPSLE poses many difficulties. The prevalence of NP symptoms in the patients with SLE varies and accounts for 21% to 95%, but only in 13% to 38% of cases these symptoms are linked to SLE.1,2 The manifestations of NPSLE have been classified into 19 syndromes that cover neurologic and psychiatric illnesses of the central, peripheral, and autonomic nervous systems.3,4 The NP events may also be classified as focal (eg, cranial neuropathy or seizure) or diffuse (eg, mood disorder, psychosis), as well as minor (headache, mild depression, anxiety, or cognitive impairment) or major (eg, cerebrovascular event, neuropathy, movement disorder, transverse myelitis, seizure, meningitis, organic brain syndrome, and psychosis).1,3,5
Most NPSLE symptoms occur at the beginning or early stage of the disease.2,6 The nervous system (primarily the central nervous system [CNS]) is one of the targets for dysregulated immune response in SLE; however, the exact pathogenesis of NPSLE is unknown.7 The possible pathomechanisms involved are thrombotic angiopathy and vasculitis.7,8
Distinguishing primary NPSLE from other neuropsychiatric conditions is challenging.9 Conventional magnetic resonance imaging (MRI) of the brain—the imaging technique of choice in the diagnostic process of NPSLE—often demonstrates nonspecific abnormalities of the white matter (from focal hyperintensities to severe large lesions) or might even not detect any pathological changes in patients with NPSLE.10,11
Therefore, various models for attribution of NP symptoms have been developed to help diagnose primary NPSLE. One of the models facilitating correct attribution of NP manifestations is the Italian model proposed by Bortoluzzi et al12 in 2015. It is based on NP symptom definitions developed by the American College of Rheumatology (ACR)3 and includes the evaluation of SLE activity, results of imaging studies, and the analysis of cerebrospinal fluid.13,14
To the best of our knowledge, there are no published studies on the attribution of NP manifestations to primary NPSLE in Polish patients that also present a detailed characterization of the disease activity and symptoms. The only published study addressing the issue of NP manifestations in Polish patients with SLE presents a description of the symptoms without their attribution.15 Therefore, in this study, we aimed to assess the prevalence of primary NPSLE using the Italian attribution model in a Polish cohort of patients with SLE. Moreover, we tried to characterize the populations of the patients with primary NPSLE and those with NP symptoms not attributed to SLE.
Patients and methods
Patients
The study included patients examined in the Department of Rheumatology and Osteoporosis, J. Struś Municipal Hospital in Poznań, Poland in the years 2015 to 2021, who fulfilled the 2012 criteria of the Systemic Lupus International Collaborating Clinics (SLICC) for SLE classification.14 All the patients included in the study signed an informed consent before the enrolment. The study was approved by the Bioethical Committee of the Poznan University of Medical Sciences (107/21).
Methodology
In this retrospective, cross‑sectional study, the following clinical information was gathered using a questionnaire:
1 Demographic data (age, sex);
2 Medical history / clinical data;
3 Current treatment;
4 Laboratory investigations: (i) titer and profile of antinuclear antibodies (ANAs); (ii) anti–double strand DNA antibodies and antiphospholipid antibodies (APLAs); (iii) concentration of complement components 3 (C3) and 4 (C4);
5 Hematologic measurements (blood count, biochemistry);
6 Disease activity measured with (i) the Systemic Lupus Erythematosus Disease Activity Inde × 2000 (SLEDAI‑2K)16,17 and (ii) the Physician Global Assessment (PGA);18
7 Organ damage measured by the SLICC/ACR Damage Index (SDI);19
8 Cognitive dysfunction assessed by neuropsychological examination with the use of the following tests: (i) spontaneous word list generation test,20 (ii) Trail Making Test, versions A and B,21 (iii) 15‑Word List Recall Test,21 (iv) Raven’s Colored Progressive Matrices,21 and an attention to detail test.22
The SLEDAI‑2K is a global index assessing lupus disease activity in the preceding 10 days. It consists of 24 weighted clinical and laboratory variables (descriptors) of 9 organ systems. The scores of the descriptors range from 1 to 8, and the total possible score for all descriptors is 105 points.17 The SLEDAI‑2K score equal to or greater than 6 points indicates the clinically active disease.
PGA is a visual analogue scale ranging from 0 to 3 points, recommended by the European League Against Rheumatism (EULAR)23 for evaluating disease activity, response to treatment, and SLE remission.18
The SDI evaluates organ damage, defined as irreversible organ dysfunction that is present for 6 months or longer, regardless of the etiology, in all organ systems. The SDI is calculated based on organ damage accumulated from SLE onset until the last visit. Its score ranges from 0 to 45 points.19
Attribution of neuropsychiatric manifestations
The NP manifestations were categorized following the 1999 ACR glossary.3 Minor and major manifestations were classified based on definitions given by Ainiala et al.1 The attribution of manifestations was performed using the Italian model developed and validated by Bortoluzzi et al.12 It comprises 4 domains: (1) the temporal relationship of NP events to the diagnosis of SLE; (2) the presence of minor or major NP events; (3) the recognition of confounding factors, and (4) the inclusion of favoring factors. The last 2 domains are based on the appendix to the 2010 EULAR recommendations for the management of SLE with NP manifestations that listed items divided into those enhancing the possibility of primary NPSLE and those decreasing those chances.24 This model comprises a simple numerical algorithm summing the points acquired in the abovementioned domains, yielding a score ranging from 0 to 10. A cutoff of 7 points or more identified primary NPSLE patients with a sensitivity of 87.9% and a specificity of 82.6%.13
Immunoassays
Immunoglobulin (Ig) G ANAs were assessed on the HEp‑2 cell line using the indirect immunofluorescence assay. Anti‑dsDNA antibodies were assessed with monospecific sandwich enzyme‑linked immunosorbent assays (ELISAs). The elevated level of anti‑dsDNA was defined as 2 times the upper limit of normal (ULN). APLA positivity was defined as positive lupus anticoagulant (dilute Russell’s viper venom time in a screening test and correction / neutralization as a confirmation test) or anti–β2‑glycoprotein‑1 in the IgG or IgM class exceeding ULN in the ELISA or anti‑cardiolipin in IgG or IgM class autoantibodies exceeding ULN in the ELISA.
Statistical analysis
Statistical analysis was conducted using Statistica v. 13.3 (TIBCO Software, Inc., Palo Alto, California, United States) and MedCalc v. 19.8 (MedCalc Software Ltd., Ostend, Belgium). Data in the Tables were presented as frequency and percentage for categorical variables or median with interquartile range for continuous variables. In the univariable analysis, the χ2 test was used to compare categorical data, and the Fisher exact test was applied when expected frequencies were small (n <40 or n <5 in one of the categories). Continuous data with nonnormal distribution were analyzed with the Mann–Whitney test. Statistical significance in our study was defined as a P value equal to or lower than 0.05.
Results
Baseline characteristics of the study group
The study included 164 patients with SLE. A total of 21 individuals were excluded from the analysis due to incomplete data; therefore, the total number of study patients was 143. Demographic characteristics, information on the disease onset, duration, symptoms, and current immunosuppressive medication in the patients with and without attributed NP manifestations are presented in Table 1. Among all the study patients, 34 (23.8%) had NP manifestations attributed to SLE. The remaining 109 patients (76.2%) had no NP manifestations or these manifestations were not attributed to SLE. The age of the patients with NPSLE and their age at the disease onset were significantly lower than in those without NP symptoms attributed to SLE (P = 0.009 and P = 0.03, respectively). The distribution of symptoms was similar in the 2 patient groups except for vasculitis, which occurred in a higher proportion of patients with primary NPSLE (P = 0.046). The patients with NPSLE were more frequently treated with immunosuppressive drugs than those without attributed NP manifestations (P = 0.02).
Parameter | Patients with NP manifestations attributed to SLE (n = 34) | Patients without NP manifestations and with NP manifestations not attributed to SLE (n = 109) | P value | ||
Data are presented as number (percentage) of patients or median (interquartile range).
Statistical significance tested by: a Fisher exact test b Mann–Whitney test c χ2 test
Abbreviations: APS, antiphospholipid syndrome; AZA, azathioprine; CsA, cyclosporin A; CQ, chloroquine; CTX, cyclophosphamide; GC, glucocorticoid; HCQ, hydroxychloroquine; IV, intravenous; MMF, mycophenolate mofetil; MTX, methotrexate; NP, neuropsychiatric; SLE, systemic lupus erythematosus | |||||
Female sex | 30 (88.2) | 102 (93.6) | 0.31a | ||
Age, y | 38 (29–45) | 45 (32–55) | 0.009b | ||
Age of disease onset, y | 35 (24–38) | 40 (25–48) | 0.03b | ||
Disease duration, y | 3.5 (2–7) | 5 (3–10) | 0.11b | ||
Symptoms | Mucocutaneous | 28 (82.3) | 94 (86.2) | 0.06a | |
Musculoskeletal | 26 (76.5) | 70 (64.2) | 0.18c | ||
Hematological | 10 (29.4) | 27 (24.8) | 0.59c | ||
Vasculitis | 7 (20.6) | 8 (7.3) | 0.046c | ||
Renal | 13 (38.2) | 38 (34.9) | 0.72c | ||
Leukopenia | 8 (23.5) | 20 (18.3) | 0.51c | ||
Thrombocytopenia | 3 (8.8) | 11 (10.1) | 0.99a | ||
APS | 2 (5.9) | 6 (1.8) | 0.93a | ||
Current use of immunosuppressive drugs | Any type | 31 (91.2) | 79 (72.4) | 0.02a | |
MMF | 7 (20.6) | 22 (20.2) | |||
AZA | 7 (20.6) | 28 (25.7) | |||
CsA | 2 (5.9) | 5 (4.6) | |||
CTX | 11 (32.4) | 10 (9.2) | |||
MTX | 4 (11.8) | 14 (12.8) | |||
Current GC use | 30 (88.2) | 82 (75.2) | 0.07a | ||
Current GC dose | ≤7.5 mg/day | 13 (38.2) | 39 (35.8) | 0.23c | |
7.5–10 mg/day | 4 (11.8) | 24 (22) | |||
>10 mg/day | 2 (5.9) | 4 (3.7) | |||
High‑dose IV pulse therapy | 11 (32.4) | 15 (13.8) | |||
Current background therapy with antimalarials (CQ/HCQ) | 29 (85.3) | 85 (80) | 0.35c | ||
Disease activity
The disease activity assessed by SLEDAI‑2K and PGA, organ damage (measured by SDI), C3/C4 levels, anti‑dsDNA antibodies concentration, and ANA profile in the patients with and without attributed NP manifestations are presented in Table 2. The disease activity measured by SLEDAI‑2K, SLEDAI‑2K after excluding NP manifestations, clinical SLEDAI‑2K, and PGA was significantly higher in the patients with NPSLE than in those without NP symptoms attributed to SLE (P = 0.001, P = 0.004, P = 0.001, and P <0.001, respectively). Consistently, the proportion of patients with the clinically active disease was higher in the group with NPSLE (P = 0.01). The SDI score did not differ between the 2 groups of patients. However, after excluding NP damage from the calculation, this index was significantly lower in the patients with NPSLE (P = 0.04). More patients with NPSLE had elevated anti‑dsDNA antibodies in the serum than those without NP manifestation or with manifestations not attributed to SLE (P = 0.01).
Parameter | Patients with NP manifestations attributed to SLE (n = 34) | Patients without NP manifestations and with NP manifestations not attributed to SLE (n = 109) | P value | ||
Data are presented as number (percentage) of patients or median (interquartile range).
a Defined as SLEDAI‑2K ≥6 points in clinical manifestations
Statistical significance tested by: b Mann–Whitney test c Fisher exact test d χ2 test
Abbreviations: AMA, antimitochondrial antibody; ANA, antinuclear antibody; APLA, antiphospholipid antibody; cSLEDAI‑2K, clinical Systemic Lupus Erythematosus Disease Activity Index version 2000 (SLEDAI‑2K without anti‑dsDNA antibodies and C3/C4 complement components); PGA, Physician Global Assessment scale; SDI, Systemic Lupus International Collaborating Clinics / American College of Rheumatology Damage Index; SLEDAI‑2K, Systemic Lupus Erythematosus Disease Activity Index version 2000; others, see Table 1 | |||||
SLEDAI‑2K, points | 26.5 (22.5–34.2) | 10 (6–16) | 0.001b | ||
Patients with clinically active diseasea | 34 (100) | 93 (85.3) | 0.01c | ||
Disease activity (SLEDAI‑2K) | Low (≤5) | 0 | 16 (14.7) | <0.001c | |
Moderate (6–10) | 0 | 46 (42.2) | |||
High (11–19) | 6 (17.6) | 29 (26.6) | |||
Very high (≥20) | 28 (83.4) | 18 (16.5) | |||
SLEDAI‑2K without NP manifestations, points | 12 (10–17.5) | 10 (6–13) | 0.004b | ||
cSLEDAI‑2K, points | 24 (18.5–30.7) | 8 (6–12) | 0.001b | ||
PGA, points | 2 (1–3) | 1 (0–2) | <0.001b | ||
SDI, points | 0 (0–1) | 0 (0–1) | 0.28b | ||
SDI without NP damage, points | 0 (0–0) | 0 (0–1) | 0.04b | ||
Patients with organ damage (SDI ≥1) | 12 (35.3) | 47 (43.1) | 0.42d | ||
Low C3/C4 level | 21 (61.8) | 55 (50.4) | 0.25d | ||
Elevated anti‑dsDNA antibodies | 27 (79.4) | 55 (50.4) | 0.01d | ||
Both low C3/C4 level and elevated anti‑dsDNA antibodies | 18 (52.9) | 40 (36.7) | 0.09d | ||
ANA titer ≥1:160 | 33 (97) | 109 (100) | 0.24c | ||
ANA profile | Anti‑Sm | 10 (29.4) | 17 (15.6) | 0.07d | |
Anti‑RNP | 5 (14.7) | 23 (21.1) | 0.42d | ||
Anti‑SSA | 17 (50) | 54 (49.5) | 0.96d | ||
Anti‑Ro52 | 14 (41.2) | 57 (52.3) | 0.30d | ||
Anti‑SSB | 6 (17.6) | 25 (22.9) | 0.51d | ||
Anti‑nucleosome | 16 (47.1) | 33 (30.3) | 0.07d | ||
Anti‑histone | 7 (20.6) | 13 (11.9) | 0.27c | ||
Anti‑rib‑P positivity | 8 (23.5) | 14 (12.8) | 0.13c | ||
AMA positivity | 2 (5.9) | 7 (6.4) | 0.98c | ||
APLA positivity | 4 (11.8) | 9 (8.3) | 0.51c | ||
Neuropsychiatric manifestations and their attribution to neuropsychiatric systemic lupus erythematosus
The overall number of patients with NP manifestations was 78 (58% of the cohort). A total of 34 patients had NP manifestations attributed to SLE (43.6% of the patients with NP symptoms), and in 44 individuals, the NP manifestations were not attributed to SLE (56.4%). The distribution of patients with NP manifestations is presented in Table 3.
NP manifestation | Total (n = 78) | Attributed to SLE (n = 34) | Not attributed to SLE (n = 44) |
Data are presented as number (percentage) of patients.
Abbreviations: CNS, central nervous system; PNS, peripheral nervous system; others, see Table 1 | |||
Solely CNS manifestations | 68 (87.2) | 25 (73.5) | 43 (97.7) |
PNS and CNS manifestations | 10 (12.8) | 9 (26.5) | 1 (2.3) |
>1 NP manifestation | 42 (53.8) | 15 (44.1) | 27 (61.4) |
Major manifestations | 47 (60.2) | 34 (100) | 13 (29.5) |
Solely minor manifestations | 31 (39.7) | 0 | 31 (70.5) |
Altogether, 155 NP manifestations were observed during the study, of which 52 (34%) were attributed to NPSLE (Table 4). In the CNS, 142 NP manifestations were observed—115 were classified as diffuse and 27 as focal (Figure 1). The NP manifestations in the peripheral nervous system (PNS) accounted for 13 cases (Table 4). Seventy‑four NP manifestations were major, and 81 were minor (Table 4).
NP manifestation | Total (n = 155) | Attributed to SLE (n = 52) | Not attributed to SLE (n = 103) |
Data are presented as number (percentage) of manifestations.
Abbreviations: see Table 1 | |||
Central nervous system | |||
Any type | 142 (91.6) | 42 (80.8) | 100 (97.1) |
Aseptic meningitis | 1 (0.6) | 1 (1.9) | 0 |
Cerebrovascular disease | 18 (11.6) | 15 (28.8) | 3 (2.9) |
Demyelinating syndrome | 2 (1.3) | 2 (3.8) | 0 |
Headache | 33 (21.3) | 2 (3.8) | 31 (30.1) |
Movement disorder (chorea) | 0 | 0 | 0 |
Myelopathy | 1 (0.6) | 1 (1.9) | 0 |
Seizure disorders | 8 (5.2) | 6 (11.5) | 2 (1.9) |
Acute confusional state | 2 (1.3) | 1 (1.9) | 1 (0.9) |
Anxiety disorder | 15 (9.7) | 1 (1.9) | 14 (13.6) |
Cognitive dysfunction | 32 (20.6) | 5 (9.6) | 27 (26.2) |
Mood disorder | 25 (16.1) | 3 (5.8) | 22 (21.3) |
Psychosis | 5 (3.2) | 5 (9.6) | 0 |
Peripheral nervous system | |||
Any type | 13 (8.4) | 10 (19.2) | 3 (2.9) |
Guillain–Barré syndrome | 1 (0.6) | 1 (1.9) | 0 |
Autonomic disorder | 1 (0.6) | 1 (1.9) | 0 |
Mononeuropathy | 2 (1.3) | 1 (1.9) | 1 (0.9) |
Myasthenia gravis | 1 (0.6) | 0 | 1 (0.9) |
Cranial neuropathy | 5 (3.2) | 5 (9.6) | 0 |
Plexopathy | 0 | 0 | 0 |
Polyneuropathy | 3 (1.9) | 2 (3.8) | 1 (0.9) |
Severity | |||
Major | 74 (47.7) | 48 (92.3) | 26 (25.2) |
Minor | 81 (52.2) | 4 (7.7) | 77 (74.7) |
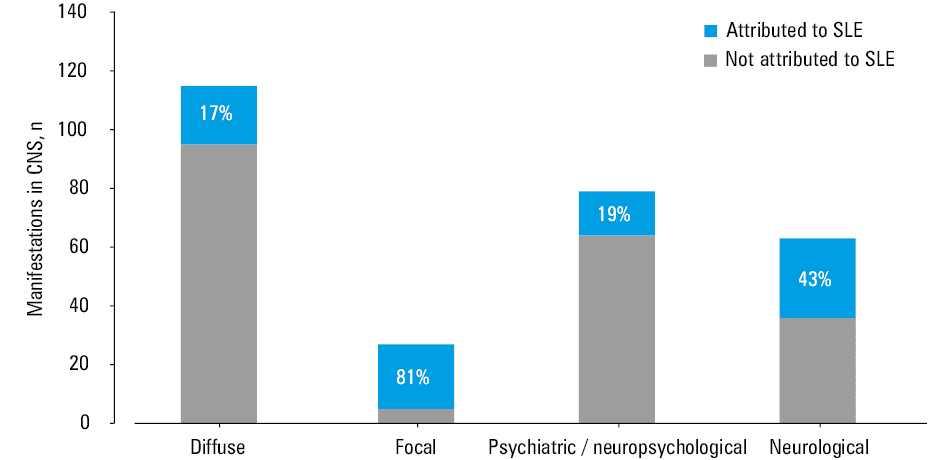
The most frequent manifestations in the CNS (as defined by the ACR in 1999) among all the patients were headache (n = 33) and cognitive dysfunction (n = 32), followed by mood disorder (n = 25), whereas the most frequent manifestations attributed to NPSLE were cerebrovascular disease (n = 15), followed by seizures (n = 6), cognitive dysfunction (n = 5), and psychosis (n = 5). In the PNS, the most frequent NP manifestations were cranial neuropathy (n = 5; all cases attributed to SLE) and polyneuropathy (n = 3; 2 cases attributed to SLE) (Table 4). Out of the 25 cases of mood disorder observed during the study, 9 were classified as major (2 of which were attributed to NPSLE) and 16 as minor (1 of which was attributed to NPSLE). Cognitive dysfunction was classified as major in 15 cases (5 of which were attributed to SLE) and minor in 17 cases.
Discussion
The aim of this study was to assess the prevalence of primary NPSLE manifestations in a Polish cohort of patients with SLE. The attribution of NP manifestations to SLE was performed using the Italian model. The clinical data, demographic characteristics, and disease activity of the patients with primary NPSLE and those with NP symptoms not attributed to SLE were also evaluated.
We showed that among the 143 patients with SLE included in the analysis, 34 (23.8%) had primary NPSLE, whereas all the patients with NP symptoms accounted for 58%. In comparison with patients without NP manifestations attributed to SLE, the patients with primary NPSLE were younger and had earlier disease onset. They had increased anti‑dsDNA concentration and presented higher disease activity (even after excluding NP manifestations) than the patients without attributed NP manifestations. The patients with primary NPSLE were also more frequently treated with immunosuppressive drugs. Without considering NP damage, the damage accrual was lower in the primary NPSLE patients than in the nonprimary NPSLE group, which might result from a younger age and earlier disease onset in the individuals with primary NPSLE. The most frequent NP manifestations in the CNS among the patients with NPSLE were cerebrovascular disease (28.8%), followed by seizures (11.5%), cognitive dysfunction (9.6%), and psychosis (9.6%). Polyneuropathy and cranial neuropathy were the sole set of NP manifestations in the PNS observed in the patients with primary NPSLE, occurring 2 (3.8%) and 5 times (9.6%), respectively.
The prevalence of primary NPSLE in published studies varies, primarily depending on the attribution model used (if any) and the race / ethnicity of studied patients. The distribution of SLE symptoms and their varying severity between populations is well known. This disease is much more frequent and has a more severe course (with higher disease activity and greater damage accrual) in non‑Caucasian populations (Hispanics, African descendants, and Asians) than in Caucasians.25
A study by Nikolopoulos et al26 on a Greek SLE cohort demonstrated slightly different proportions, although consistent with those observed in our study: 17.6% of 555 patients with SLE were diagnosed with primary NPSLE, whereas 38.4% developed at least a single NP symptom.26 In the same study, the authors identified neuropsychiatric disease as an emerging SLE phenotype in Caucasians. The proportion of patients with primary NPSLE observed in our study (23.8%) supports the conclusion of the abovementioned analysis. In contrast to Caucasian descent, the Asian race / ethnicity has been indicated as a predictor of lower risk for NP events in SLE.27 Interestingly, when a large group of over 1800 patients with SLE of variable ethnicity was analyzed, the proportions of patients experiencing NP symptoms (52.3%) and those with primary NPSLE were comparable with our results (13.5% or 21.2%, depending on the attribution model applied).28 A study on a Chinese cohort of 194 patients with NPSLE demonstrated a different distribution of NP symptoms, the most frequently occurring ones being seizures (36.6%), followed by acute confusional state (25.3%), cerebrovascular disease (15.5%), and headache (13.9%).29 Similarly to our study (as well as to the studies by other authors),30,31 the disease activity in the Chinese patients with SLE was higher in the NPSLE group than in individuals without NP symptoms (SLEDAI‑2K, mean [SD], 25.3 [8.8] vs 8.6 [6.7]).29 Seizures were also the most common (45.8%) NP syndrome in the NPSLE cohort from South Africa, followed by psychosis (18.1%) and cerebrovascular disease (18.1%).32
The higher disease activity in the patients with primary NPSLE (measured by the SELENA‑SLEDAI score) was demonstrated in a study on a Swiss SLE cohort. In this analysis, the patients with NPSLE also showed greater organ damage assessed by the SDI scale. The most frequent NP symptoms were cerebrovascular disease (25.4%), headache (26.4%), seizures (19.2%), and mood disorders (5.8%), with the last value reaching 8.8% in our study. However, the authors of the study did not distinguish between primary and secondary NP events, and the NPSLE group included all patients with SLE presenting NP symptoms.30
The diagnostic process of NPSLE is challenging since it is extremely difficult to distinguish primary NPSLE syndromes from NP manifestations not associated with SLE but with complications secondary to the SLE course or its treatment.33 The biomarkers of primary NPSLE that could help diagnose or guide therapeutic decisions do not yet exist.34 Because of these problems, different models for attribution of NP events to SLE have been developed. The efforts are also being made to improve the diagnostics of NPSLE with the application of MRI, a method of choice for evaluating patients with SLE experiencing NP syndromes.24
A retrospective analysis of Italian patients with SLE showed that conventional brain MRI presents nonspecific alterations, such as white matter hyperintensities and, less often, cerebral atrophy in patients with newly diagnosed NPSLE (48%), non‑NPSLE (31.8%), and even in those without any NP symptoms (20%). The NP symptoms in that study were attributed to SLE using the Italian model, as in our analysis. Importantly, a greater degree of alterations on subsequent MRI scans correlated with the occurrence of new NP events.35 A study on a Dutch cohort showed a higher white matter volume on MRI in individuals with NPSLE than in non‑NPSLE patients. Structural changes in the brain observed on MRI also differed between 2 phenotypes of NPSLE: inflammatory and ischemic. The white matter and total brain volume were lower in the patients with NPSLE experiencing an inflammatory phenotype than in those with an ischemic phenotype.31 Higher volume of the left and right caudate nuclei was also associated with the disease activity (measured by SLEDAI‑2K) in the patients with NPSLE.36 Advanced MRI techniques may convey more subtle insights on brain alterations, for example, the severity of depression and anxiety in the patients with NPSLE was associated with hemodynamic and functional connectivity changes of the frontolimbic neural circuit.37
The correct diagnosis of NPSLE is of great importance, as neuropsychiatric involvement at SLE onset is associated with transition to the severe form of the disease. Moreover, mortality among the patients with NPSLE has been observed to be higher than in the SLE patients without NP manifestations.28,38 However, a study on a smaller cohort using a different attribution model of NP manifestations demonstrated that the mortality among the patients with major NPSLE did not differ from the mortality of those without NP symptoms and those with minor NPSLE.39 That study categorized SLE patients based not only on NP symptoms’ presence but also on the treatment: major NPSLE was defined as a disease requiring immunosuppressive or anticoagulant therapy, whereas minor NPSLE together with non‑NPSLE, as a disease where symptomatic therapy is sufficient.39
As mentioned before, several attribution models are used in the published reports; therefore, a direct comparison of the obtained results is not always possible or appropriate. Moreover, each model only approximates the diagnosis but cannot be considered a fully reliable tool for the correct attribution of the NP symptoms.30 Therefore, employing the Italian model as a scientific tool may be considered a limitation of this study, although this is a current practice in the field. The number of patients analyzed was not high, which also counts as a weakness of this study. Nonetheless, the observed prevalence of primary NPSLE and frequencies of NP symptoms are generally in line with other published data. Thus, even with this number of participants, we could observe the actual distribution of NP manifestations.
In conclusion, the presented study is the first one to aim for the correct attribution of NP events in a Polish cohort of patients with SLE. Primary NP manifestations occurred mainly in young patients with high disease activity. Cerebrovascular disease, seizures, psychosis, cognitive dysfunction, and cranial neuropathy were the most frequent manifestations of NPSLE. In light of the debilitating character of NP events and a considerable proportion of SLE patients with primary NPSLE, further studies are required to identify better diagnostic tools and treatment options.
- Ainiala H, Loukkola J, Peltola J, et al. The prevalence of neuropsychiatric syndromes in systemic lupus erythematosus. Neurology. 2001; 57: 496‑500. | Crossref
- Tay SH, Mak A. Diagnosing and attributing neuropsychiatric events to systemic lupus erythematosus: time to untie the Gordian knot? Rheumatology (Oxford). 2017; 1: i14‑i23. | Crossref
- ACR ad hoc Committee on Neuropsychiatric Lupus Nomenclature. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999; 42: 599‑608. | Crossref
- Mackay M. Lupus brain fog: a biologic perspective on cognitive impairment, depression, and fatigue in systemic lupus erythematosus. Immunol Res. 2015; 63: 26‑37. | Crossref
- Govoni M, Bortoluzzi A, Padovan M, et al. The diagnosis and clinical management of the neuropsychiatric manifestations of lupus. J Autoimmun. 2016; 74: 41‑72. | Crossref
ARTICLE INFORMATION