Nephropathic cystinosis in Poland: a 40-year retrospective study
Key words: clinical course, epidemiology, genetics, nephropathic cystinosis



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Nephropathic cystinosis in Poland: a 40-year retrospective study
Introduction: Nephropathic cystinosis (NC) is a rare, autosomal recessive disorder leading to lysosomal accumulation of cystine. It is caused by mutations in the CTNS gene encoding a cystine cotransporter cystinosin. The infantile (INC) and juvenile (JNC) forms are distinguished. The former, responsible for 95% of cases, is characterized by development of renal Fanconi syndrome, end‑stage kidney disease (ESKD), and extrarenal complications. A therapy with cysteamine significantly improves outcomes. There are limited data on NC in the Central Eastern European countries.
Objectives: We aimed to evaluate the prevalence, genetic background, and clinical course of NC in the Polish population.
Patients and methods: We performed a retrospective analysis of data of all identified NC patients in Poland.
Results: Between 1982 and 2017, 15 patients with NC (13 ICN, 2 JCN) were identified. The most common mutations of the CTNS gene were c.18_c.21delGACT and c.681+1G>A, whereas only 2 patients carried the 57 kb deletion. The majority (11/13) of INC patients with limited access to the cysteamine therapy developed ESKD at a median age of 11 years and 9 of them received kidney transplants. Three INC patients died at a median age of 24 years. In contrast, 2 INC patients treated adequately present normal kidney function and growth at the age of 13 and 11 years. Two JNC patients presented a milder course.
Conclusions: The prevalence of NC in Poland is much lower than in the Western countries and its molecular background appears to be different. The unfavorable course in the majority of INC patients was caused by a limited access to the cysteamine treatment.
What's new?
This is the first comprehensive report on nephropathic cystinosis (NC) from Poland. To date, the prevalence, clinical course, genetic background, treatment, and outcome of this rare inherited metabolic disease was unknown in Poland. The results of this study indicate that the prevalence of NC in Poland is much lower than in the Western countries, and also its molecular background appears to be different. The latter is based on infrequent occurrence of the common 57 kb deletion, and higher proportion of other pathogenic variants of the CTNS gene, mainly c.18_c.21delGACT and c.681+1G>A. It should be noted that an unfavorable course in the majority of Polish patients was caused by a historically limited accessibility of the specific cystine depletion therapy with cysteamine. Fortunately, better access to diagnostic and therapeutic procedures has significantly improved patient care in recent years.
Introduction
Cystinosis is a rare (estimated prevalence of 1:150 000), autosomal recessive disorder leading to systemic lysosomal accumulation and crystallization of cystine, an oxidized dimer of the amino acid cysteine. It is caused by biallelic pathogenic variants of the CTNS gene (17p13) encoding the lysosomal transmembrane cystine‑proton cotransporter cystinosin.1-3 Depending on the severity of kidney disease and on the age of onset, the disorder has 2 nephropathic and 1 non‑nephropathic form.1 The most common and severe form, responsible for almost 95% of cases, is infantile NC (INC; Online Mendelian Inheritance in Man [OMIM] #219800) appearing in infancy and accompanied by renal Fanconi syndrome (FS), growth retardation, ocular complications with photophobia, rapidly progressive chronic kidney disease (CKD) within the first decade of life, and final extrarenal multiorgan damage. Patients of the European origin usually have blond hair, white skin, and blue eyes.1,4 Juvenile nephropathic cystinosis (JNC; OMIM #219900) accounts for approximately 5% of cases and represents a significantly milder and usually nonspecific phenotype, including isolated proteinuria or incomplete FS (iFS), slow progressive CKD, and late‑onset extrarenal complications including ocular involvement.1,5,6 Casuistic non‑nephropathic disease, known as the ocular / adult form (OMIM #219750) is limited to corneal cystine crystals deposition without systemic involvement.7 The clinical diagnosis of NC is usually based on the characteristic phenotype and confirmed by elevated cystine level in white blood cells (WBC) and molecular analysis.1,8,9 To date, over 160 pathogenic variants of the CTNS gene have been identified. The 57 kb deletion encompassing the complete CTNS gene and the SHPK gene is by far the most common and has been detected in almost 50% of the affected individuals of the Northern European descent.10
The cystine depletion therapy with cysteamine, commercially available for the last 30 years, significantly improved the quality of life and outcomes for NC patients.11,12 The drug reacts with cystine within lysosomes and enables a mixed disulfide to exit into the cytoplasm through a cationic amino acid transporter (SLC66A1).13 The treatment prevents or limits the majority of cystinosis‑related complications and significantly delays the progression of CKD.1,11,14,15
There are no official data on cystinosis prevalence in the Central Eastern European countries and only 2 reports, focused on ocular complications in a series of Polish and Hungarian patients have been published.16,17 Therefore, we systematically evaluated the prevalence, clinical course, genetic background, treatment, and outcome of patients with cystinosis in Poland.
Patients and methods
A retrospective analysis of available data of all identified Polish patients with NC reported to the Polish Registry of Inherited Tubulopathies (POLtube) was performed. This working group of the Polish Society for Pediatric Nephrology, established in 2012, constantly collects data of Polish patients with selected, genetically determined kidney disorders. To ensure completeness of the reported NC cases, an additional survey was distributed among all 15 Polish centers of pediatric nephrology. As most identified Polish NC patients reached the adulthood, 2 adult medical centers which followed them participated in the study. A dedicated chart review, distributed in a survey form, comprised selected anthropometrical, clinical, and laboratory parameters, including serum creatinine, WBC cystine levels, and genetic testing results obtained throughout individual observation periods.
The estimated glomerular filtration rate (eGFR) was calculated in pediatric patients using the original or modified Schwartz equation,18,19 and in adults using the Modification of Diet in Renal Disease formula.20 Available WBC cystine levels were initially measured by foreign laboratories (ie, Department of Pediatric Kidney and Metabolic Diseases, Medical School of Hannover, Germany), and since 2014 at the Department of Biochemistry, Medical University of Gdańsk, Poland. The results were expressed in nmol ½ cystine/mg protein. Where available, molecular genetic analyses for the CTNS gene were performed at the Department of Pediatrics, University of Münster, Germany and at the Institute of Human Genetics, University of Cologne, Germany by the targeted Sanger sequencing, and in selected cases by the Multiplex Ligation‑dependent Probe Amplification (MLPA) with a commercially available MLPA kit (P473 CTNS from MRC Holland, Amsterdam, the Netherlands) for copy number analysis.
Statistical analysis
Statistical analysis was performed using STATISTICA 13.1 software (StatSoft, Tulsa, Oklahoma, United States). Data are presented as the median (range) as appropriate for continuous variables and as absolute numbers and / or percentages for categorical variables. As the study was based on retrospective data collected via the POLtube between 2012 and 2021, additional Local Ethics Committee approvals were not applied for. Written informed consents were obtained from patients or patients’ guardians if applicable for the publication of images.
Results
The study comprised 15 individuals with NC from 14 families diagnosed over a 35‑year period between 1982 and 2017. There were 13 (87%; 5 boys and 8 girls) INC and 2 (13%) JNC patients (both girls). All their parents were Caucasians and nonconsanguineous. The patient clinical characteristics are summarized in Table 1 and shown in detail in Supplementary material, Table S1.
Parameter | INC (n = 13) | JNC (n = 2) | ||
Data are presented as number (percentage) or median (range).
Abbreviations: CNS, central nervous system; eGFR, estimated glomerular filtration rate; ESKD, end‑stage kidney disease; FS, Fanconi syndrome; FU, follow‑up; iFS, incomplete Fanconi syndrome; INC, infantile nephropathic cystinosis; JNC, juvenile nephropathic cystinosis; KT, kidney transplant; KTx, kidney transplantation; WBC, white blood cells | ||||
Age at the last FU, y | 30.7 (10.8–38.3) | 21.2 (16.8–25.7) | ||
Duration of FU, y | 29.7 (10–37.3) | 9.8 (7–12.7) | ||
Age at first clinical symptoms, y | 1 (0.5–1.5) | 5.7 (2–9.5) | ||
First clinical symptoms | Failure to thrive | 12 (92) | 0 | |
Motoric retardation | 12 (92) | 0 | ||
Rickets | 9 (69) | 0 | ||
Polyuria / polydipsia | 8 (62) | 0 | ||
Recurrent vomiting | 5 (38) | 0 | ||
FS | 8 (62) | 0 | ||
iFS | 5 (38) | 0 | ||
Isolated proteinuria | 0 | 2 (100) | ||
Hypercalciuria | 6 (46) | 2 (100) | ||
Decreased eGFR | 5 (38) | 1 (50) | ||
Bilateral hyperechogenicity of renal cortex | 3 (23) | 2 (100) | ||
Age at clinical diagnosis, y | 2 (0.5–30) | 16 (12–20) | ||
Time between first symptoms and clinical diagnosis, y | 1 (0.1–29) | 4.7 (2.5–18) | ||
WBC cystine measurement | 11 (85) | 2 (100) | ||
Age at the first WBC cystine measurement, y | 28 (0.5–33) | 16 (12–20) | ||
First WBC cystine level, nmol ½ cystine/mg protein | 4.54 (1.2–18) | 2.28 (2.04–2.53) | ||
Molecular evaluation | 11 (85) | 2 (100) | ||
Age at molecular evaluation, y | 26 (5–35) | 12 (11–13) | ||
Regular systemic cysteamine treatment during FU | 2 (15) | 0 | ||
ESKD during FU | 11 (85) | 0 | ||
Age at ESKD, y | 11 (6–15) | – | ||
Kidney transplantation during FU | 9/11 (82) | 0 | ||
First KTx | 9/11 (82) | – | ||
Age at the first KTx, y | 12 (6–16) | – | ||
Duration of the first KT function, y | 16 (6–22.5) | – | ||
Loss of the first KT | 3/9 (33.3) | – | ||
Second KTx after loss of the first KT | 3/9 (33) | – | ||
Age at the second KTx, y | 23 (19–31) | – | ||
Duration of the second KT function, y | 11 (7.3–13) | – | ||
Extrarenal symptoms during FU | Any type | 13 (100) | 2 (100) | |
Ocular | 13 (100) | 2 (100) | ||
Primary hypothyroidism | 13 (100) | 0 | ||
Short stature | 10 (77) | 0 | ||
Diabetes mellitus | 6 (46) | 0 | ||
Dysphagia | 4 (31) | 0 | ||
Distal myopathy | 3 (23) | 0 | ||
CNS involvement | 3 (23) | 0 | ||
Mortality during FU | 3 (23) | 0 | ||
Age at death, y | 24 (12–36) | – | ||
Clinical manifestation of infantile nephropathic cystinosis
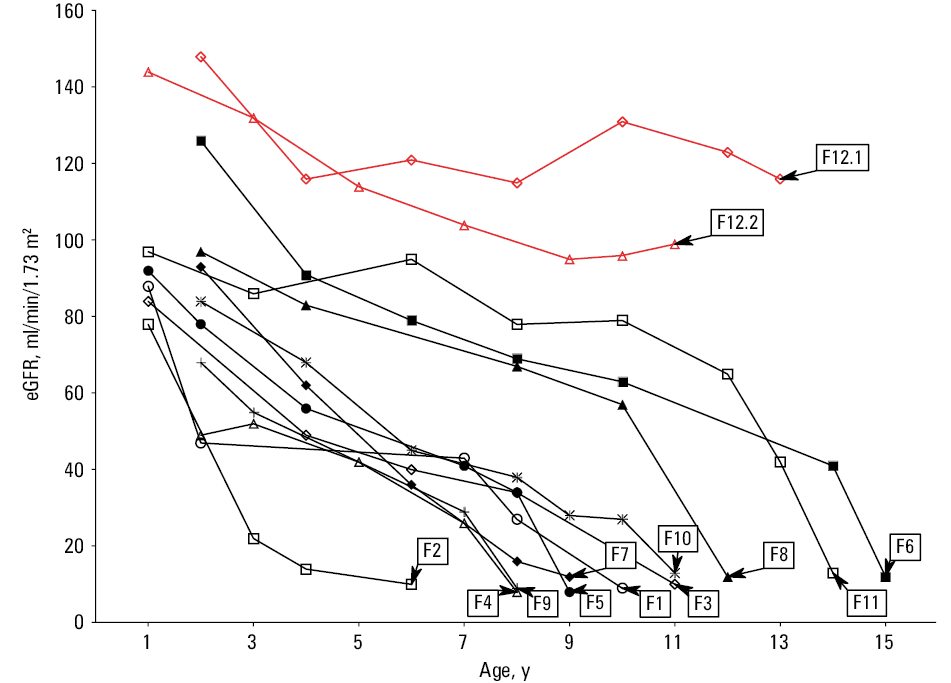
The median age at the last follow‑up of INC patients was 30.7 (10.8–38.3) years. Median age at the onset of manifestations was 1 (0.5–1.5) year and the median time of the follow‑up from the onset was 29.7 (10–37.3) years. The distribution of initial symptoms was as follows: failure to thrive (92%), motoric retardation (92%), rickets (69%), polyuria / polydipsia (62%), and recurrent vomiting (38%). They were accompanied by complete (62%) or incomplete FS (38%) and hypercalciuria (HC) (46%). Decreased eGFR was initially present in 38% of the patients, while 23% of the patients were suspected of nephrocalcinosis when examined due to increased bilateral echogenicity of the renal cortex.
The median (range) age at clinical diagnosis of INC was 2 (0.5–30) years and it was made 0.1–29 years after the initial symptom onset. An exception was the patient F2 who was initially treated as for chronic tubulointerstitial nephritis of unknown etiology, and subsequently diagnosed correctly at 30 years of age. In the majority of patients, initial diagnosis of INC was made due to the characteristic phenotype, comprising FS and corneal cystine crystals deposits. Subsequently, it was confirmed in 11 out of 13 patients by elevated WBC cystine level, although 7 patients had a long‑standing delay. The initial median WBC cystine level was 4.54 (1.2–18) nmol ½ cystine/mg protein (normal <0.2 nmol ½ cystine/mg protein). WBC cystine level was not assessed in the patients F5 and F10 due to limited access to testing and / or poor parental or patient compliance. Apart from the patients F12.1 and F12.2, WBC cystine levels were assessed only occasionally.
Molecular results of infantile nephropathic cystinosis
Molecular evaluation was performed in 11 out of 13 INC patients and pathogenic variants in the CTNS gene were revealed in 9 of them. Of those evaluated, 4 patients were compound‑heterozygotes, and 3 homozygotes for their pathogenic variant. In 2 patients, mutations were only detected in 1 CTNS allele. No mutations were found in the patient F6, while the patient F3 was suspected of homozygous deletion of exon 3. The most frequent pathogenic variant was the c.18_c.21delGACT (T7Ffs*7) deletion. It affected 4 independent patients, of whom 2 were homozygous (F8, F9), 1 (F4) compound‑heterozygous (c.681+1G>A; c.18_c.21delGACT [p.T7Ffs*7]) and 1 (F1) had only 1 allele affected. The second most common was the intronic mutation c.681+1G>A. It affected 3 independent patients of whom 1 (F11) was homozygous, 1 (F4) heterozygous, and in 1 (F5) it was detected in a single allele. Two patients (sisters F12.1 and F12.2) were compound heterozygous for the 57 kb deletion + c.314_317del4 (p.fs166X). In the patient F2 a novel splice mutation c.141–1 G>A was identified.
Therapy of infantile nephropathic cystinosis
Oral cysteamine (Cystagon) treatment was started early enough in only 2 patients (F12.1 and F12.2), at the age of 1.5 and 0.6 years, respectively, and it was continued adequately during the follow‑up. In the remaining patients, this treatment was initiated after their third year of life and was conducted transiently or intermittently, mainly due to the limited access. In all cases, a symptomatic treatment of FS with potassium, bicarbonates, and phosphate supplementation was introduced but its adequacy during the follow‑up could only be confirmed in patients F12.1 and F12.2.
Kidney failure in infantile nephropathic cystinosis patients
During follow‑up, 11 out of 13 patients with INC developed ESKD at the median (range) age of 11 (6–16) years, even though the course of CKD was variable (Figure 1). The patients 12.1 and 12.2, currently at the age of 13 and 11 years, show normal kidney function with eGFR of 116 and 97 ml/min/1.73 m2, respectively. Of those patients that developed ESKD, 9 underwent kidney transplantation (KTx) at the median (range) age of 12 (6–16) years. The median (IQR) time of kidney graft function was 16 (6–22.5) years and only 3 patients (F1, F2, and F4) developed terminal graft failure during this follow‑up. Subsequently, the latter 3 patients underwent retransplantation at the median age of 23 (19–31) years. Currently, 2 of them present either normal (F5) or deteriorated (F2) graft function, while the patient F1 died with a functioning graft and the eGFR of 45 ml/min/1.73 m2.
Extrarenal complications of infantile nephropathic cystinosis
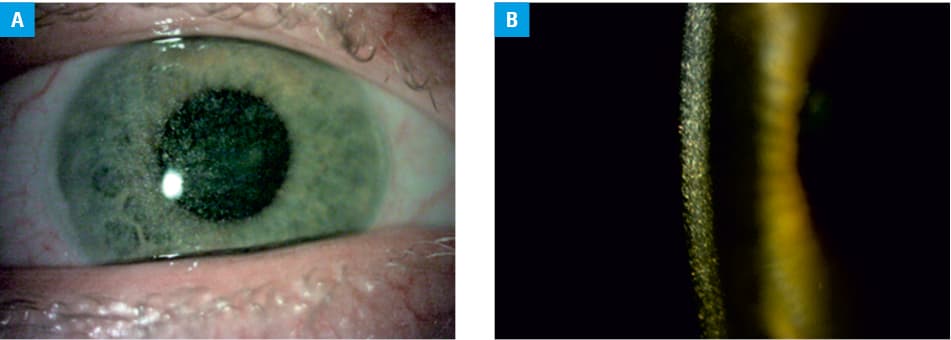
All the patients developed some extrarenal complications of NC. During the follow‑up, all of them showed ocular cystine crystal deposits of variable degree and the subsequent clinical consequences (Figure 2). All patients suffered from photophobia and 3 of them (F2–F4) required keratoplasty due to serious keratopathy. Despite this procedure, the patient F4 developed unilateral blindness. Only the patients 12.1 and 12.2 received early and continuous treatment with eye drops containing cysteamine. The treatment began with an aqueous formulation of 0.1% cysteamine hydrochloride at the age of 3 and 2 years, respectively, and continued with commercial Cystadrops following its official approval by the European Medicines Agency 4 years ago. Most remaining patients received this treatment inconsistently. A detailed ophthalmological status of 5 of these patients (F2, F4, F11, F12.1, and F12.2) was reported previously.16
All INC patients developed mild primary hypothyroidism in childhood and supplementary therapy with L‑thyroxin was provided in all cases.
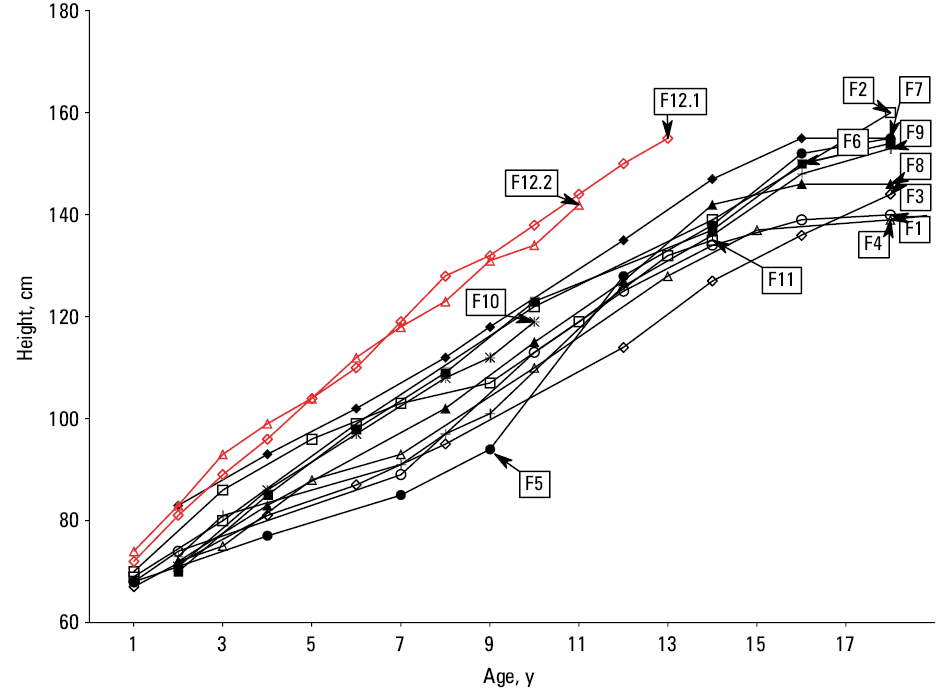
Of the 13 INC patients, 10 presented with short stature (<3rd percentile) with the final median (IQR) adult height of 153 (139–160) cm (Figure 3). Some of them (F3, F7, F8, F11) were treated with recombinant human growth hormone (rhGH) during adolescence, however, it was only successful in 1 patient (F7). rhGH was not used in patients 12.1 and 12.2, however, they showed normal growth at 25–50 and 10–25 percentiles at the current age of 13 and 11 years, respectively.
Dysphagia was present in 4 out of 13 (F1, F4, F7, F10) patients, including 1 pediatric case (F10), and 3 out of 13 patients (F1, F2, F4) showed brain involvement (nonspecific demyelination, cortical atrophy, epilepsy, and migraine) and distal myopathy. Diabetes mellitus (DM) developed in 6 out of 13 patients.
Overall, 3 out of 13 INC patients died during the follow‑up. The patient F1 died at 36 years due to myocardial infarction, the patient F9 at the age of 24 years from cerebral stroke, and the pediatric patient F10 at 12 years due to fatal accidental choking.
Two women (F4, F7) got pregnant at the age of 29 years (20 years after KTx) and 27 years (8 years after KTx), and delivered healthy children at 33 and 32 gestational weeks, respectively.
Characterization of juvenile nephropathic cystinosis patients
So far, only 2 women (F13, F14) with JNC were identified. Their age at the latest follow‑up was 16.8 and 25.7 years, respectively. At onset, both patients presented with isolated proteinuria and hypercalciuria at the age of 2 and 9 years. The patient F13 was initially treated as for nutcracker syndrome, although after a kidney biopsy at 13 years of age, a focal segmental glomerulosclerosis was also considered. At the age of 20, ophthalmological evaluation revealed corneal cystine crystals.16 The final diagnosis was confirmed by increased WBC ½ cystine level (2.53 nmol ½ cystine/mg protein) and compound heterozygous mutations in the CTNS gene (c.225+5_c.225+6delGT/insCC; c.530A>G(N177S). Currently, she still presents with proteinuria without FS and her eGFR is slightly decreased at 87 ml/min/1.73 m2 at the age of 26. Due to photophobia, she receives topical treatment with cysteamine (Cystadrops) and does not currently present other extrarenal symptoms.
Following a kidney biopsy at the age of 10, the patient F14 was initially treated as for chronic tubulointerstitial nephritis of unknown origin. However, at 12 years of age ophthalmological evaluation revealed corneal cystine deposits. Increased WBC ½ cystine level (2.04 nmol ½ cystine/mg protein) and compound heterozygous mutations of the CTNS gene (c.629T>C [p.Leu210Pro];c.462‑27_462‑3del) confirmed their final diagnosis. According to the literature, both are seemingly novel variants.10 At the age of 17 years, she still presents with proteinuria without other signs of FS and her eGFR is slightly decreased at 72 ml/min/1.73 m2. Since the age of 16, she has received regular systemic and topical treatment with cysteamine (Cystagon, Cystadrops), and except for mild ocular involvement,16 she has no other extrarenal symptoms.
Discussion
The precise worldwide epidemiology of cystinosis has not been established, nevertheless the national surveys from several Western countries estimate its incidence at approximately 0.5 to 1 per 100 000 live births.1,21 It has been also suggested that it may be higher in the Middle East, Turkey, Pakistan, and North Africa, where consanguineous marriages are more frequent.9,10,21 To date, strictly epidemiological data from the Central Eastern and Eastern European countries are not available, and except for 2 limited ophthalmological studies from Poland and Hungary,16,17 only single cases of NC from this region (Russia, Serbia, and Czech Republic) have been reported so far.22-24 In our study, we were able to identify 15 Polish NC patients who were diagnosed over a 35‑year period between 1982 and 2017. In relation to the current Polish population of about 38 million people, this number suggests a much lower cystinosis incidence in Poland than in the Western countries. Interestingly, we have recently found a similar epidemiological discrepancy for primary hyperoxaluria, another very rare monogenic disorder leading to ESKD.25
The reason for low NC incidence in the Polish population is unclear. Theoretically, the carrier frequency of pathogenic CTNS variants in the ethnic Polish population could be lower than in the Western countries. Other reasons may be related to the low incidence of consanguineous marriages and limited emigration from regions of high disease burden. Interestingly, the 57 kb deletion accounted only for 18% of the analyzed cases and 10% of the families in our cohort. This common variant is also rare in patients from Italy, East Mediterranean, and the Middle East.9 It has been speculated that the 57 kb deletion represents a founder effect and appeared in the territory of today’s Germany prior to AD 700, spreading afterwards throughout Western and Northern Europe via migration of Germanic and Viking people.26 As these migrants very rarely settled in the Central Eastern Europe, we hypothesize that it may explain the rarity of this mutation among Polish patients. The difference in the genetic background of Polish patients with INC and those of the Northern European origin is seen in the spectrum of pathogenic variants of the CTNS gene in our cohort. The most common mutations were c.18_c.21delGACT (T7Ffs*7) deletion and intronic mutations (c.681+1G>A), affecting 4 out of 11 (36%) and 3 out of 11 (27%) patients, respectively. The former is a widespread variant, reported in patients from many countries (United Kingdom, France, the Netherlands, Italy, France, Spain, Thailand, and Iran) but most frequently from Turkey, accounting for 14% of the mutant alleles.10,27,28 The second most common mutation in our group (c.681+1G>A) was already detected in patients from the United Kingdom and Italy.10 Interestingly, mutations c.225+5_c.225+6delGT/insCC and c.530A>G (p.N177S) identified in the patient F13 with JNC phenotype were found only in single patients with INC from Saudi Arabia, United States, and Australia. This provides new insights into the discussion on genotype‑phenotype correlations in cystinosis.
The most severe renal consequence of untreated INC is the development of ESKD, which occurs at a median age of 9 years.29 In some cases, deterioration of GFR is already seen before the second year of life,30 which is similar to our cohort (38% of the patients).
The outcome of KTx in the patients with cystinosis is usually favorable.31 It has also been observed in our cohort, as the median duration of the first kidney graft function was 16 years. Three retransplanted patients (F1, F2, F7) maintained their second grafts for a median of 11 years of the follow‑up. This supports the hypothesis that cystinosis may act as a protective factor for kidney graft survival, potentially due to a better immune tolerance as a result of the mTORC1 pathway downregulation.32
Chronologically, the eye is the second organ affected by symptoms of cystine accumulation. In our patients, corneal cystine deposits caused photophobia at a median age of 3 to 4 years. Subsequently, a majority of ocular tissues are affected and adult patients may suffer from glare disability, decreased contrast sensitivity, stinging, epiphora, glaucoma, blepharospasm, vision impairment, and even blindness due to band keratopathy.33 Most of these findings were observed in our cohort and have been recently described in detail.16
The majority of other extrarenal complications of INC occur in adulthood and progress with age. Even after a successful KTx, the patients remain at a risk of late endocrinopathies, myopathies, or neuropathies, as well as cardiovascular, gastrointestinal, and pulmonary problems.1,4 As in other studies, the most common endocrine finding in our cohort was primary hypothyroidism, following its subclinical form.4 Pancreatic involvement frequently leads to glucose intolerance and finally to overt DM, which may be exacerbated by maintenance immunosuppressives after KTx (46% in our cohort).4,34 A male primary hypogonadism leading to infertility is another irreversible endocrine complication.35 In contrast, women with cystinosis are usually fertile and may deliver healthy children, as evidenced by 2 patients in our cohort.36 Some patients (23% in our cohort) developed a progressive myopathy with distal muscle wasting and weakness. Myopathies may also manifest as dysphagia (31% in our cohort), which may lead to aspiratory pneumonias.37 Although dysphagia belongs to the late complications of cystinosis, it was also observed in our pediatric patient (F10), who died due to fatal choking at the age of 12 years. Cystinosis patients may experience a broad spectrum of central neurological complications (28% in our cohort). Vascular calcification is a predominant problem among cardiovascular complications, which may lead to an early myocardial infarction, as in our patient F1.38 This risk is compounded by CKD, hypertension, and hypercholesterolemia, which are frequently observed in cystinotic patients.39
Severe growth impairment is a characteristic feature of the natural course of INC, as the patients generally do not grow above the third percentile and rarely reach 150 cm in adulthood.1,40 The pathological mechanism is complex and includes the consequences of FS, CKD, coexisting endocrinopathies, cystine deposition in bone tissue, suspected cystinosis‑related intrinsic bone defect, and lack of cystinosin.41 In our cohort, growth deficit was present in 77% of the cases.
Maintenance cystine depletion therapy delays or prevents the majority of multiorgan complications, especially when introduced before the age of 1.5–2 years.1,4,12 In particular, it may delay CKD progression to ESKD by approximately 10 years.14,15,28 The response to treatment must be regularly monitored by WBC cystine level.42 Of note, the treatment efficacy is often limited by nonadherence. This is likely due to unpleasant taste and halitosis, gastrointestinal discomfort, and strict dosage regimen. It was assumed that despite “milder” course of nephropathy and other complications, the patients with JNC may also benefit from this therapy.6 Importantly, cysteamine does not treat FS, therefore its main metabolic consequences must be treated symptomatically to prevent severe clinical symptoms, including rickets.43 It has been previously demonstrated that early introduced, systemic cystine depletion therapy accompanied by effective symptomatic treatment of FS results in significantly better linear growth.12 rhGH therapy is regarded as effective management in the patients with INC,44 and is dedicated to those in whom growth deficit persists despite adequate cysteamine therapy and sufficient metabolic control.41 We observed that rhGH therapy was not satisfactory in 75% of patients from our cohort, likely due to its delayed initiation, short duration, and insufficient treatment of the primary disease, which confirms the importance of the combined long‑term management.
Although kidney replacement therapies significantly improved the outcome of INC patients, a median survival time shortly before the era of systemic cysteamine treatment was only 8.5 years.31,45 Subsequent studies showed a decreasing mortality rate, especially if cystine depletion therapy was started early and was continued throughout life. This was mainly related to later onset of ESKD and alleviation of extrarenal complications.14,15 As a result, the current life expectancy of INC patients may extend past 50 years.45 Unfortunately, due to the previous nonavailability of cysteamine 11 out of 13 Polish INC patients developed ESKD at the median age of 11 years, presented severe growth retardation, and other extrarenal complications. In contrast, 2 patients (F12.1, F12.2) from our cohort, who were diagnosed during the era of drug reimbursement, show normal kidney function and physical development (currently) at the age of 13 and 11 years. Of note, in Poland the drug program for INC treatment with Cystagon was approved by the Minister of Health and has been financed by the National Health Fund only since 2014.
The limitations of our study are related to its retrospective nature, which might lead to the information bias. In particular, we could not completely exclude the fact that some Polish NC patients remained un- or misdiagnosed or have been underreported leading to an underestimation of NC prevalence in Poland. However, we strongly believe that multicenter and longitudinal patient data collection by the POLtube registry, characteristic clinical course of NC, and significantly improved awareness of NC among Polish pediatric nephrologists minimized this possibility. In addition, we were not able to perform MLPA analysis in selected cases to improve the detection rate of pathogenic variants of the CTNS gene.
Conclusions
To the best of our knowledge, this is the first comprehensive study on NC from the Central Eastern European region. Our results indicate lower incidence of this disease in Poland in comparison with the Western countries. Infrequent occurrence of the 57 kb deletion and higher proportion of the other pathogenic variants of the CTNS gene, mainly c.18_c.21delGACT and c.681+1G>A, suggest different molecular background in the Polish INC cohort. The limited availability of the cystine depletion therapy was a major problem in the past and has negatively influenced the outcome of the majority of Polish patients during the 35 years for which our cohort has been compiled. Although there are still some systemic barriers, including a deficit of adult metabolic physicians or a lack of a systematic transition from pediatric into adult health care, raising awareness among professionals and better access to diagnostic and therapeutic procedures significantly improved patient care in the recent years.
- Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med. 2002; 347: 111‑121. | Crossref
- Town M, Jean G, Cherqui S, et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet. 1998; 18: 319‑324. | Crossref
- Kalatzis V, Cherqui S, Antignac C, Gasnier B. Cystinosin, the protein defective in cystinosis, is a H(+)-driven lysosomal cystine transporter. EMBO J. 2001; 20: 5940‑5949. | Crossref
- Nesterova G, Gahl W. Nephropathic cystinosis: late complications of a multisystemic disease. Pediatr Nephrol. 2008; 23: 863‑878. | Crossref
- Goldman H, Scriver CR, Aaron K et al. Adolescent cystinosis: comparisons with infantile and adult forms. Pediatrics. 1971; 47: 979‑988. | Crossref
SUPPLEMENTARY MATERIAL
ARTICLE INFORMATION