Ataxia denotes impaired balance and coordination of gait, posture, limbs, and speech. It usually results from disturbances of the cerebellum and its connections (cerebellar ataxia) or sensory pathways (sensory ataxia). In most patients, it is secondary to toxic (alcohol, antiepileptics), vascular (stroke), immune (paraneoplastic, multiple sclerosis), infectious (SARS‑CoV‑2, HIV, cerebellar abscess), and neoplastic (metastatic or, more rarely, primary tumor) causes.1-3 Genetic ataxias include spinocerebellar ataxias (SCAs), with an average estimated prevalence worldwide of up to 5.6 per 100 000.1 There are more than 40 distinct forms of SCA, among which the most common is SCA type 3 (SCA3; also referred to as Machado–Joseph disease), accounting for 20% to 50% of all SCA cases.1,4
SCA3 is a progressive neurodegenerative disease first described 50 years ago in 2 families (Machado and Joseph) of Portuguese‑Azorean descent.4 It is caused by abnormal repeat expansion in the ATXN3 gene. Healthy individuals have up to 44 cytosine‑adenine‑guanine (CAG) repeats, whereas those who develop SCA3 usually have more than 60 repeats.1,4 The mutation causes the expression of aberrant protein with expanded polyglutamine stretch that easily aggregates and deposits in the neurons of the cerebellum and brainstem.1,4 This leads to the typical clinical presentation of progressive cerebellar ataxia and worsening of speech. Magnetic resonance imaging of the brain reveals atrophy of the brainstem and cerebellum (Figure 1). Some patients may also manifest other motor symptoms, including extrapyramidal syndromes (basal ganglia involvement), amyotrophy (brainstem and spinal cord involvement), and neuropathy (peripheral nervous system involvement).4 Autonomic symptoms, particularly bladder dysfunction (nocturia and urinary incontinence), sudomotor (abnormal thermoregulation, cold intolerance, hypohidrosis), and cardiovascular disturbances (orthostatic hypotension), affect the majority of patients.4,5 Autonomic cardiac denervation is mostly parasympathetic and occurs in up to 60% of patients; however, sympathetic dysfunction was also demonstrated with iodine‑123 metaiodobenzylguanidine cardiac scintigraphy.5 Psychiatric features, predominantly depressive symptoms, affect up to 60% of SCA3 patients, and their basis (organic vs reactive) is not yet understood.5 Cognitive impairment, particularly worsened learning abilities and memory dysfunction, was reported in many patients.5 Fatigue occurs in up to 61% of cases, and its severity makes it one of the most disabling symptoms in SCA3.5 Chronic pain, particularly in the lumbosacral region, and painful muscle cramps affect up to 80% of patients.4,5 The latter may be the first manifestation of the disease even in a quarter of patients.5 Sleep disturbances are common and include REM sleep behavior disorder, restless legs syndrome, sleep apnea, insomnia, and daytime sleepiness.5
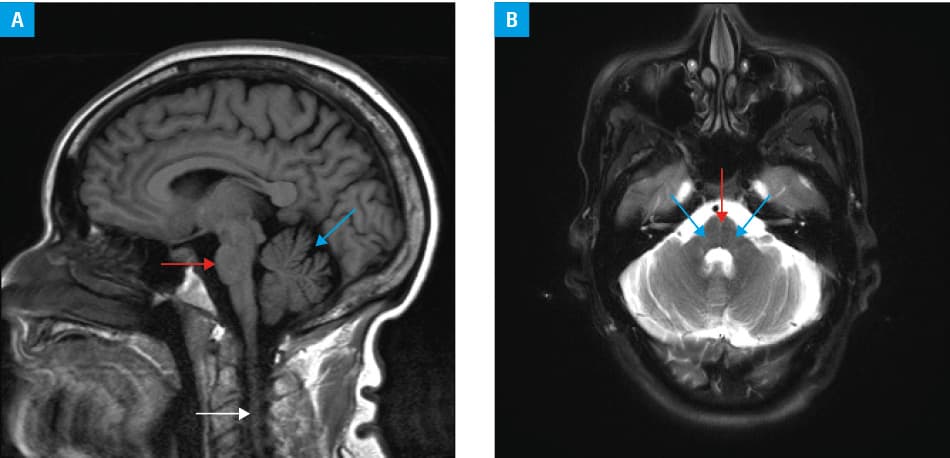
Currently, there is no approved therapy for SCA3.1,4 The preliminary reports suggest beneficial effects of riluzole in SCA3 and other SCAs.1,4 The therapeutic effect of riluzole is attributed to its antiglutamatergic activity in the brain.1,4 Of note, close monitoring for adverse effects, particularly during the first year of treatment, is necessary, as some patients develop liver toxicity ranging from mild elevation of aminotransferases to liver failure. Troriluzole, a prodrug of riluzole with a better safety profile and promising efficacy in SCA3, is in phase III of a clinical trial (NCT03701399).
- Klockgether T, Mariotti C, Paulson HL. Spinocerebellar ataxia. Nat Rev Dis Primers. 2019; 5: 24. | Crossref
- Adamczyk‑Sowa M, Niedziela N, Kubicka‑Bączyk K, et al. Neurological symptoms as a clinical manifestation of coronavirus disease 2019: implications for internists. Pol Arch Intern Med. 2021; 131: 54‑62. | Crossref
- Hirschfeld AS. Autoimmune mediated hyperkinetic movement disorders in SARS‑CoV‑2 infection ‑ a systematic review. Neurol Neurochir Pol. 2021; 55: 549‑558. | Crossref
- Paulson H, Shakkottai V. Spinocerebellar Ataxia Type 3. In: Adam MP, Mirzaa GM, Pagon RA, et al., eds. GeneReviews. Seattle (WA): University of Washington, Seattle; Copyright 1993‑2022, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.; 1993.
- Moro A, Moscovich M, Farah M, et al. Nonmotor symptoms in spinocerebellar ataxias (SCAs). Cerebellum Ataxias. 2019; 6: 12. | Crossref
ARTICLE INFORMATION