Endocrine paraneoplastic hypercalcemia in a patient with pancreatic neuroendocrine neoplasm



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Endocrine paraneoplastic hypercalcemia in a patient with pancreatic neuroendocrine neoplasm
Endocrine paraneoplastic hypercalcemia occurs in up to 10% of patients with neoplasms, and the most common cause is the tumor secretion of parathyroid hormone–related peptide (PTHrP).1 Neuroendocrine neoplasms (NENs) may produce several types of peptides, however, paraneoplastic hypercalcemia in NENs is exceedingly rare.1-4
In October 2018, a 35‑year‑old white woman with NEN‑associated severe hypercalcemia was referred to our endocrinology department. A month earlier she had been admitted to another hospital due to an acute confusional state. At that time, she reported poor appetite, occasional nausea, vomiting, indigestion, and a 5 kg weight loss in the last 12 months. Gastroscopy and brain computed tomography (CT) were unremarkable. However, ultrasound of the abdomen showed numerous hyperechoic, solid formations in the liver and a hypoechoic, solid lesion in the tail of the pancreas. Total body CT scan confirmed the presence of multiple metastases, up to 40 mm in diameter, in both lobes of the liver and a lesion 20 mm in diameter in the pancreatic tail. Moreover, enlarged lymph nodes with a maximum diameter of 18 mm in the left subdiaphragmatic area were found. Histopathologic examination of the liver biopsy specimen revealed well‑differentiated NEN, Ki67 inde × 10%. Immunohistochemistry was positive for CD56, synaptophysin, panCK, and chromogranin A. Laboratory evaluation showed hypercalcemia (4.92 mmol/l; reference range [RR], 2.15–2.5 mmol/l) and acutely raised serum creatinine (254.3 mmol/l; RR, 44–80 mmol/l). Treatment with intravenous saline, loop diuretics, corticosteroids, and bisphosphonates was introduced. When the patient arrived at our facility, biochemical tests of calcium metabolism showed persistent hypercalcemia (3.87 mmol/l), hypophosphatemia (1.7 mg/dl; RR, 2.3–4.7 mg/dl), increased activity of total alkaline phosphatase (315 U/l; RR, 37–98 U/l) and its bone‑type isoenzyme (83.4 μg/l; RR, 3–19 μg/l), a low level of 25‑hydroxyvitamin D (18.6 ng/ml; RR, 20–80 ng/ml) with a normal level of 1,25‑dihydroxyvitamin D (28.40 pg/ml; RR, 25–86.5 pg/ml), and low parathyroid hormone level (<3 pg/ml; RR, 10–62 pg/ml). Based on the results, we suspected PTHrP‑related hypercalcemia, which was confirmed by a markedly elevated serum concentration of PTHrP (38 pmol/l; RR, <1.3 pmol/l). Moreover, we found a high level of chromogranin A (193.2 ng/ml; RR, <84.7 ng/ml), slightly elevated gastrin level (123 pg/ml; RR, 13–115 pg/ml), and normal level of serotonin (227 nmol/l; RR, 138–1080 nmol/l). Somatostatin receptor scintigraphy revealed strong expression of somatostatin receptors in the primary tumor and metastases (Krenning score 4). We continued the therapy of hypercalcemia with intravenous isotonic saline, furosemide, corticosteroids, and bisphosphonates, and initiated treatment with short- and long‑acting somatostatin analogs (SSA) (Figure 1A). Due to insufficient control of hypercalcemia despite the treatment, we administered 4 cycles of peptide receptor radionuclide therapy (PRRT) with a lutetium‑177 (177Lu)-based probe linked to the somatostatin analog octreotate ([177Lu]Lu‑DOTA‑TATE) and referred the patient for a distal pancreatectomy and splenectomy with simultaneous radiofrequency ablation of the liver metastases (Figure 1A). Histopathologic examination of the primary lesion of the pancreatic tail showed NEN grade 2 according to the 2019 World Health Organization grading system with Ki67 inde × 15%.4 After the treatment, we observed improvement in the clinical condition, normalization of calcium, phosphorus, and parathyroid hormone (PTH) levels, full recovery of renal function, and reduction of the PTHrP level (Figure 1A). Moreover, follow‑up CT scans showed a marked reduction in the tumor mass in the liver and withdrawal of adenopathy (Figure 1B and 1C). After 22 months of the disease stability on SSAs only, we observed a recurrence of symptoms, hypercalcemia (Figure 1D), increase of the PTHrP level, and tumor progression (Figure 1E). The patient was started on chemotherapy with temozolomide and capecitabine (CAPTEM), with satisfactory control of her metastatic disease (Figure 1D and 1F). She remains under the care of an endocrinology and oncology outpatient clinic. Despite the CAPTEM and long‑acting SSA therapy, she takes vitamin D3 in a prophylactic dose of 2000 IU daily, metformin due to pancreatogenic diabetes, and timonacic for metastatic liver disease.
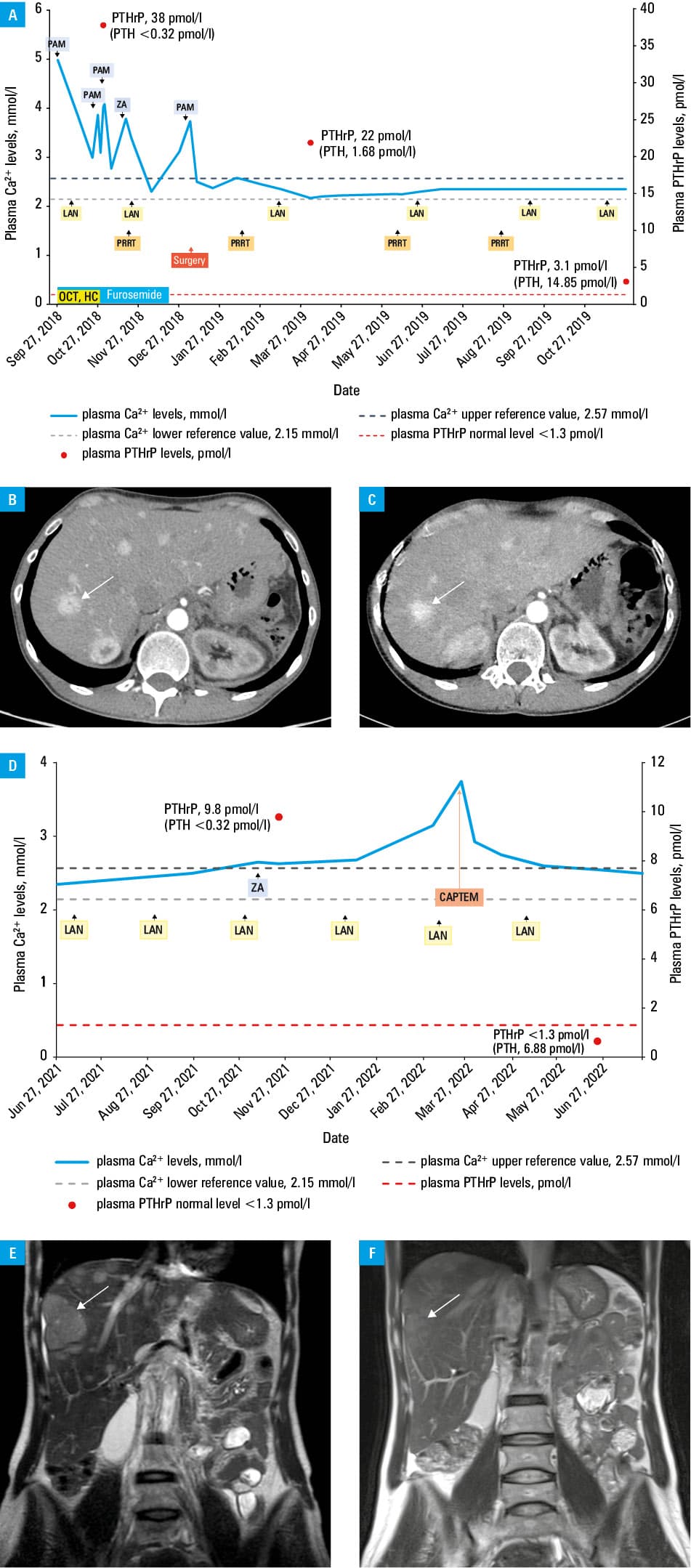
Abbreviations: CAPTEM, temozolomide and capecitabine; LAN, lanreotide; PAM, pamidronate; PRRT, peptide receptor radionuclide therapy; PTH, parathyroid hormone; ZA, zoledronic acid
Hypercalcemia of malignancy may be due to bone metastases or paraneoplastic secretion of humoral factors. According to the literature, pancreatic NENs producing PTHrP are the most common cause of humoral hypercalcemia in NENs.1,2 Ectopic secretion of PTH or 1,25‑dihydroxyvitamin D by NENs has also been reported.1 It should be highlighted that hypercalcemia associated with NENs may also result from primary hyperparathyroidism as a manifestation of multiple endocrine neoplasia type 1 syndrome.3,4 Therefore, a complete evaluation of calcium metabolism in NEN patients with hypercalcemia is mandatory. Emergency management of NENs paraneoplastic hypercalcemia should focus on lowering calcium levels. Based on a literature review, long‑term control can only be achieved by the treatment of the underlying tumor, specifically surgery, when possible, SSAs, and PRRT with [177Lu]Lu‑DOTA‑TATE.2 Regarding second‑line treatment, CAPTEM chemotherapy appears to be an effective approach, which may prolong survival in patients who have a well‑differentiated, metastatic NEN and who have progressed on previous therapies.5 Subsequent measurements of the hormone produced by NEN after therapeutic intervention with radiologic testing allow for determining the response to the therapy or the tumor recurrence.3,4
- Giannetta E, Sesti F, Modica R, et al. Case report: unmasking hypercalcemia in patients with neuroendocrine neoplasms. Experience from six Italian referral centers. Front Endocrinol (Lausanne). 2021; 12: 1‑7. | Crossref
- Kamp K, Feelders RA, Van Adrichem RCS, et al. Parathyroid hormone‑related peptide (PTHrP) secretion by gastroenteropancreatic neuroendocrine tumors (GEP‑NETs): clinical features, diagnosis, management, and follow‑up. J Clin Endocrinol Metab. 2014; 99: 3060‑3069. | Crossref
- Halfdanarson TR, Strosberg JR, Tang L, et al. The North American Neuroendocrine Tumor Society consensus guidelines for surveillance and medical management of pancreatic neuroendocrine tumors. Pancreas. 2020; 49: 863‑881. | Crossref
- Kos‑Kudła B, Foltyn W, Malczewska A, et al. Update of the diagnostic and therapeutic guidelines for gastro‑entero‑pancreatic neuroendocrine neoplasms [in Polish]. Endokrynol Pol. 2022; 73: 387‑454. | Crossref
- Milanesi A, Yu R, Wolin EM. Humoral hypercalcemia of malignancy caused by parathyroid hormone‑related peptide‑secreting neuroendocrine tumors: report of six cases. Pancreatology. 2013; 13: 324‑326. | Crossref
ARTICLE INFORMATION