Pulmonary fibrosis in primary Sjögren syndrome: computed tomography, clinical features, and associated clinical factors
Key words: computed tomography, dry cough, fibrosis, shortness of breath, Sjögren syndrome



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Pulmonary fibrosis in primary Sjögren syndrome: computed tomography, clinical features, and associated clinical factors
Introduction: Interstitial lung disease (ILD) is common in patients with primary Sjögren syndrome (pSS). Pulmonary fibrosis significantly reduces the patient’s quality of life. Therefore, better understanding of the characteristics of patients with pulmonary fibrosis is necessary.
Objectives: The aim of the study was to evaluate the computed tomography (CT) and clinical features of pSS‑related ILD and to explore the factors associated with fibrotic ILD in patients with pSS.
Patients and methods: In total, 151 patients with pSS‑related ILD were retrospectively studied for their demographic and clinical characteristics. The patients were categorized into the nonfibrosing ILD and fibrosing ILD groups based on their CT findings. The clinicl, laboratory, and CT findings from both groups were compared to determine the influencing factors associated with the condition.
Results: The nonfibrosing ILD and fibrosing ILD groups comprised 103 and 48 patients, respectively. As compared with the nonfibrosing ILD group, the fibrosing ILD group had a shorter disease duration, higher frequency of dry cough and shortness of breath, more patients with ground‑glass opacity, mediastinal lymph node disease, and pleural lesions on chest CT, and lower frequency of dry mouth and eyes. Dry cough and shortness of breath were independent predictors of pulmonary fibrosis in the patients with pSS (P = 0.01 and P = 0.02, respectively).
Conclusions: The presence of dry cough and shortness of breath in the patients with pSS may indicate concomitant pulmonary fibrosis. High‑resolution chest CT can be used for better insights on the occurrence and severity of pulmonary fibrosis in these patients.
What's new?
Pulmonary fibrosis may occur concomitantly with primary Sjögren syndrome (pSS). By comparing retrospective data of patients with and without pulmonary fibrosis, we were able to elucidate and better understand the clinical data and factors related to pulmonary fibrosis in patients with pSS. Among other variables, we found that the patients with pSS and pulmonary fibrosis had a shorter disease course, higher frequency of dry cough and shortness of breath, and lower frequency of dry mouth and eyes. Dry cough and shortness of breath were independent predictors of pulmonary fibrosis in the patients with pSS. These findings are relevant to the clinical practice of pulmonology and rheumatology and to physicians working in related specialties who may encounter patients with pSS. In the future, this information could lead to improved clinical practice guidelines for evaluating and taking care of patients with pSS.
Introduction
Primary Sjögren syndrome (pSS) is a chronic inflammatory disease of the autoimmune system often associated with symptoms of ocular and / or oral dryness and of high incidence in menopausal women aged approximately 50 years.1 Furthermore, pSS affects both the salivary and lacrimal glands as well as other parts of the body, including the lungs, skin, kidneys, and nervous system.2 Interstitial lung disease (ILD) is a manifestation of pulmonary involvement in pSS.3 ILD is characterized by diffuse damage to the lung parenchyma (mainly the interstitium).4,5 Repeated damage to this tissue can lead to pulmonary fibrosis, with a pathological process that includes fibroblast proliferation, collagen deposition, and lung structural remodeling.6 Pulmonary fibrosis is irreversible, resulting in a progressive loss of lung function.5 Therefore, understanding the early stages of pulmonary fibrosis in ILD is important for managing patients with pSS.
Numerous studies have suggested that the risk factors for Sjögren syndrome‑related ILD include age, smoking status, male sex, antinuclear antibody (ANA) positivity, rheumatoid factor (RF) level, C‑reactive protein (CRP) level, respiratory symptoms, postmenopausal period in women, low albumin levels, Raynaud syndrome, lymphopenia, and rampant caries.7-11 Our recent findings suggest that pSS patients with positive anti‑Ro52 antibodies have a lower risk of developing pulmonary fibrosis than those with negative anti‑Ro52 antibodies (odds ratio [OR], 0.419; 95% CI, 0.183–0.963).12 Based on previous findings in patients with pSS and lung involvement, we hypothesized that dry cough and shortness of breath are associated with fibrotic ILD in pSS. To test this hypothesis, we selected only patients with pSS who also had ILD to investigate the relationship between chest computed tomography (CT) features, dry cough, shortness of breath, and other clinical symptoms of pSS‑associated fibrosing ILD.
Patients and methods
Ethical considerations
All procedures in this retrospective study were conducted in adherence to the Declaration of Helsinki. The local institutional review board of Tongji Hospital (Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, China) approved the study (TJ‑IRB20180615) and waived the need for written informed consent owing to the study’s retrospective design.
Patients
We screened 1013 inpatients with SS treated in the rheumatology or respiratory medicine department of our hospital between 2012 and 2019. The screening was conducted in strict accordance with the International Classification Criteria13 or the American‑European Consensus Group SS classification criteria (Supplementary material, Tables S1 and S2).14 Overall, 214 patients with secondary SS, 83 without chest CT, and 565 without CT changes in ILD were excluded. Finally, the study included 151 patients with pSS‑ILD.
Clinical data collection
The patients’ medical records upon initial hospitalization were sourced for demographic information and laboratory data. The latter included the presence of ANA and anti‑Ro52, anti–Sjögren‑syndrome‑related antigen B (anti‑SSB), and anti–Sjögren‑syndrome‑related antigen A (anti‑SSA) antibodies, levels of RF (reference range [RR], 0–30 IU/ml), complement C3 (RR, 0.8–1.8 g/l), complement C4 (RR, 0.1–0.4 g/l), CRP level (RR, 0–10 mg/l), and white blood cell (WBC) count (RR, 3.5–10 × 109/l).
Imaging features and evaluation
Chest imaging data of 151 patients were retrieved from our hospital’s Radiology Information System workstation. The CT images were independently evaluated by 2 thoracic radiologists with extensive diagnostic experience who were blinded to the patients’ medical data and diagnoses. When the radiologists’ opinions did not align, a discussion was held, after which a consensus was reached.
Using the Fleischner Society White Paper as a reference,15 we categorized the patients into the nonfibrosing ILD and fibrosing ILD groups based on the absence or presence of reticular patterns (including honeycomb shadows) on CT images, respectively. The 2 lung lobes were divided into the outer (the most superficial), middle, and inner layers and evaluated for reticular patterns, micronodules, ground‑glass opacity, consolidation, bronchial wall thickening, interlobular septal thickening, bronchiectasis, pulmonary cysts, emphysema, mosaic perfusion, subpleural lines, pericardial effusion, mediastinal lymphadenopathy, and pleural lesions.
Statistical analysis
SPSS version 20 (IBM Corp., Armonk, New York, United States) was used for all the analyses. Data normality was assessed with the Kolmogorov–Smirnov test. Non‑normally and normally distributed variables were tested using the Mann–Whitney test, and independent sample t test, and the results are presented as medians with interquartile range (IQR) and means (SD), respectively. The categorical values were tested using the Fisher exact test or the χ2 test, as appropriate.
Potential factors influencing pulmonary fibrosis in patients with pSS were analyzed using multivariable logistic regression. First, continuous variables were considered categorical variables. WBC count and levels of C3 and C4 were categorized as decreased, normal, or increased, whereas CRP and RF levels were classified as normal or abnormal. Disease duration was divided into 2 groups: 0–24 months and above 24 months. The patients were assigned to 3 age groups: above 60 years, 40–60 years, and below 40 years old. Second, potential risk factors were screened using the univariable regression analysis. Finally, a multivariable logistic regression model composed of the variables considered clinically important and the variables with a P value below 0.1 in the univariable analysis was constructed. Considering the limited number of variables available, those included were carefully selected to ensure the simplicity of the final multivariable logistic regression model. The P value below 0.05 was considered significant (2‑tailed).
Results
The mean (SD) age of the 151 patients with pSS‑ILD (129 women, 22 men) was 53 (11.3) years. Of these patients, 48 (31.8%) and 103 (68.2%) were categorized into the fibrosing ILD and nonfibrosing ILD groups, respectively. The fibrosing ILD group had a higher prevalence of men (P = 0.047) and a more pronounced frequency of dry cough, shortness of breath, and fever (P <0.001). Additionally, the fibrosing ILD group had a shorter median duration of the illness and lower frequency of rampant caries, dry mouth, and dry eyes (P values, 0.01, 0.02, <0.001, and 0.001, respectively). The differences between the 2 groups were not significant in terms of age, dyspnea, chest pain, and frequency of Raynaud syndrome (Table 1).
Parameter | Nonfibrosing ILD group (n = 103) | Fibrosing ILD group (n = 48) | P value | |
Data are presented as number (percentage) of patients unless indicated otherwise.
Abbreviations: ILD, interstitial lung disease; IQR, interquartile range | ||||
Disease duration, mo, median (IQR) | 48 (24–102) | 30 (8–96) | 0.01 | |
Age at disease onset, y, mean (SD) | 52.56 (11.8) | 54.04 (10.03) | 0.45 | |
Sex | Women | 92 (89.3) | 37 (77.1) | 0.047 |
Men | 11 (10.7) | 11 (22.9) | ||
Dry cough | 22 (21.4) | 30 (62.5) | <0.001 | |
Expectoration | 10 (9.7) | 8 (16.7) | 0.22 | |
Chest tightness | 12 (11.7) | 7 (14.6) | 0.61 | |
Dyspnea | 3 (2.9) | 4 (8.3) | 0.14 | |
Chest pain | 5 (4.9) | 4 (8.3) | 0.47 | |
Shortness of breath | 15 (14.6) | 22 (45.8) | <0.001 | |
Fever | 15 (14.6) | 21 (43.8) | <0.001 | |
Oral dryness | 78 (75.7) | 22 (45.8) | <0.001 | |
Ocular dryness | 63 (61.2) | 16 (33.3) | 0.001 | |
Rampant caries | 22 (21.4) | 3 (6.3) | 0.02 | |
Raynaud syndrome | 12 (11.7) | 6 (12.5) | 0.88 | |
The fibrosing ILD group had higher WBC counts, levels of C3 and CRP, and a lower frequency of positive anti‑Ro52 and anti‑SSA antibodies (P values <0.001, 0.04, <0.001, <0.001, and 0.01, respectively). The C4 and RF levels and ANA and anti‑SSB antibody positivity rates in the 2 groups did not differ significantly (Figure 1).
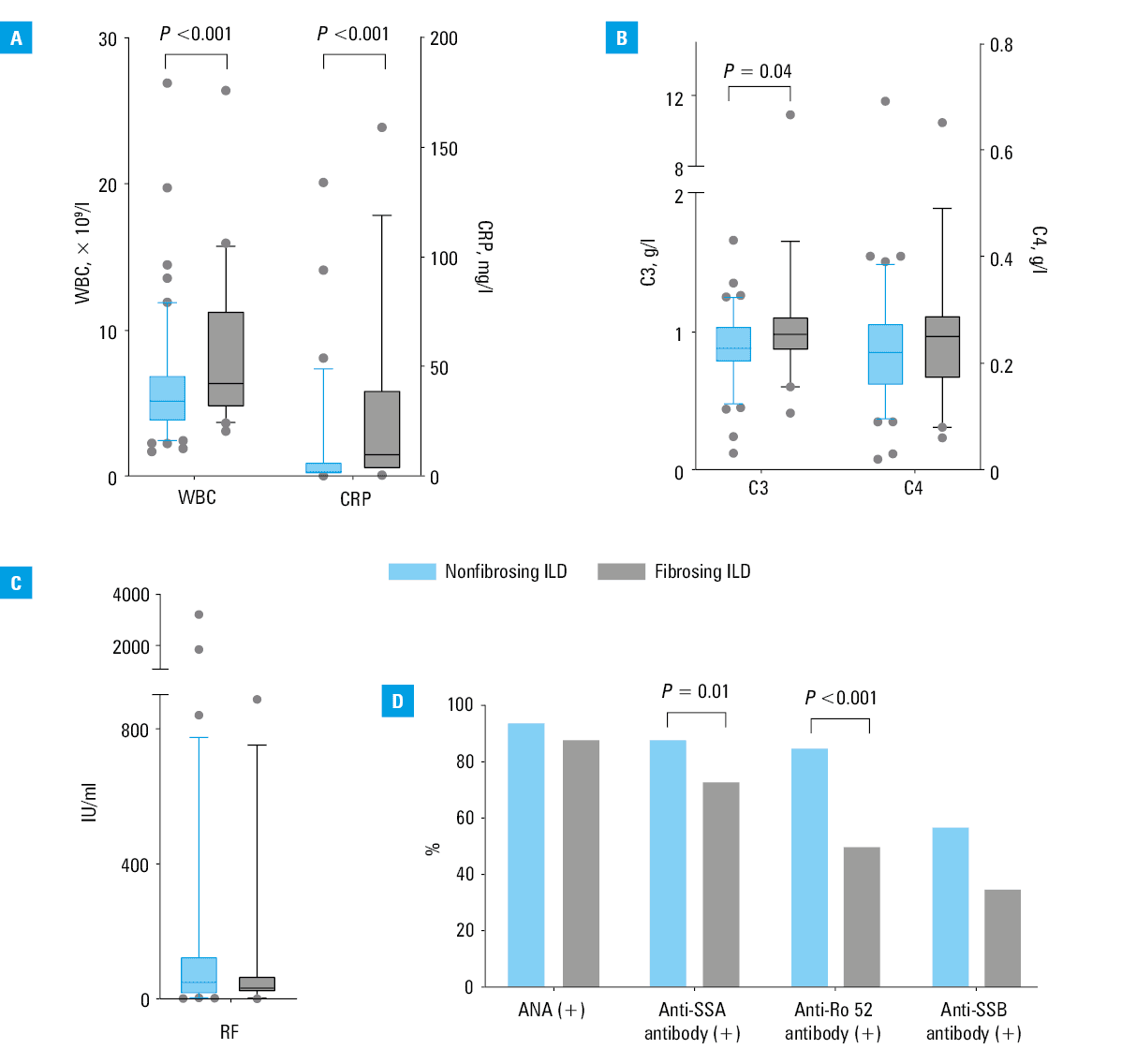
Abbreviations: see Table 1
Typically, chest CT scans of patients with pSS and pulmonary fibrosis were characterized by asymmetrical abnormalities distributed in the basal and lateral regions of the lower lobes of both lungs (Figure 2). Reticular structures consistent with pulmonary fibrosis were observed in 48 patients. The fibrosing ILD group comprised significantly more patients with ground‑glass opacities, mediastinal lymphadenopathy, bronchial wall thickening, interlobular septal thickening, subpleural lines, bronchiectasis, consolidation, and pleural thickening (P values <0.001, 0.02, <0.001, <0.001, 0.04, 0.003, 0.02, and <0.001, respectively), and fewer patients with micronodules (P = 0.001). No significant differences were observed in the frequencies of pulmonary cysts, emphysema, mosaic perfusion, pericardial effusion, and pleural effusion between the 2 groups (Figure 3).
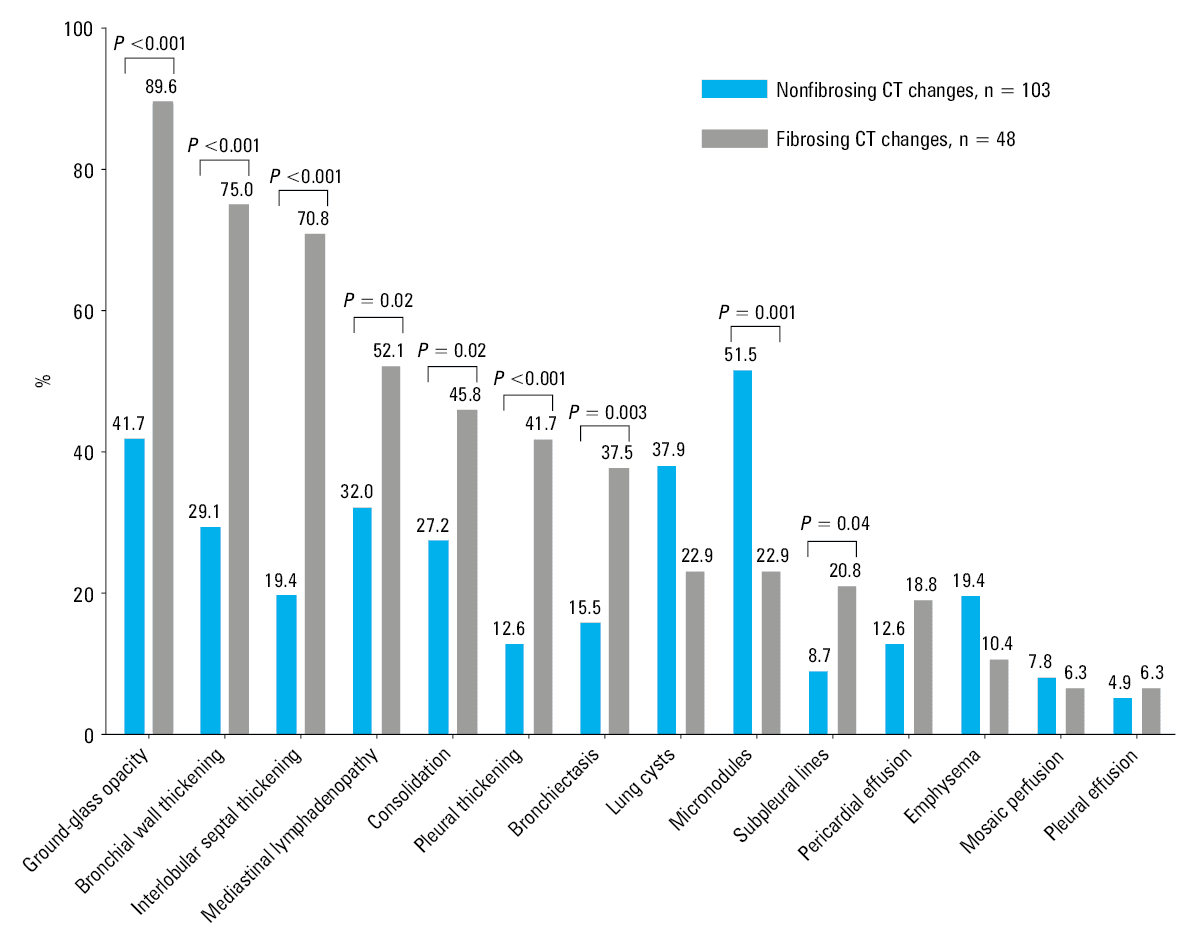
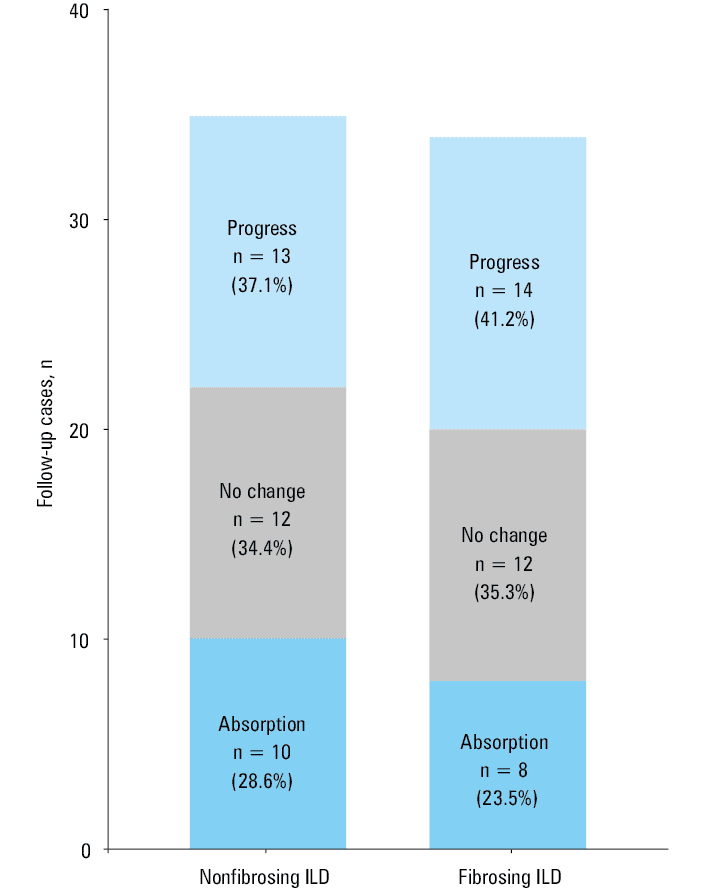
Among 151 patients with pSS‑ILD, 69 were followed up (including 34 and 35 with and without pulmonary fibrosis, respectively) for a median time of 11.5 (IQR, 4–16) months. These patients underwent a mean of 2 chest CT examinations during the study period (including CT examination at the first hospitalization), and most of them received immunosuppressive therapy (cyclophosphamide or leflunomide) and glucocorticoid therapy (prednisone acetate, meprednisone, or dexamethasone). During the treatment, the CT images showed no significant differences between the groups regarding the final changes (Figure 4).
After adjusting for confounding factors, including sex, disease duration, and age, the multivariable regression analysis revealed that dry cough and shortness of breath were independent predictive factors of pulmonary fibrosis in patients with pSS (P values 0.01 and 0.02, respectively) (Figure 5).
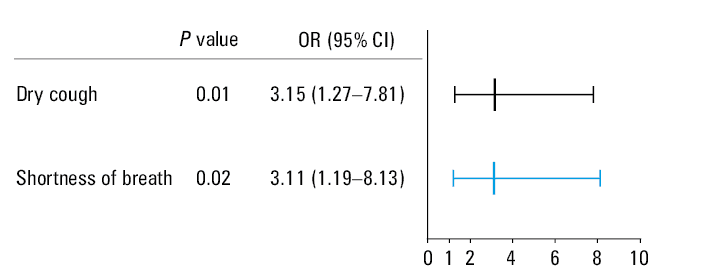
Abbreviations: OR, odds ratio
Discussion
ILD is fairly common in pSS.16-19 Patients with pSS who exhibit lung involvement have a reduced quality of life and increased mortality.20 Early detection and active treatment are important to improve prognosis. Therefore, we studied a cohort of 151 patients with pSS‑ILD in our hospital to evaluate imaging, laboratory, and clinical factors that influenced pulmonary fibrosis.
We found that shortness of breath and dry cough were the main symptoms of pSS‑ILD. Dry cough and shortness of breath were more common in the fibrosing ILD group and were independent predictive factors of pulmonary fibrosis among patients with pSS. A previous study16 also indicated dry cough as one of the factors related to pSS‑ILD. Dry cough occurs in approximately 41% to 61% of patients with SS.21 This may be attributed to dry airways with condensed secretions, bronchial hyper‑responsiveness, inflammation of the bronchi and bronchioles, abnormal mucociliary clearance, and gastroesophageal reflux.22 Furthermore, the severity of the cough may be associated with tracheal dryness (localized SS).22 However, the severity of ILD does not necessarily correlate with the severity of the extrapulmonary manifestations of pSS, and the signs of airway involvement may be minimal or absent.23 Based on the prevalence of dry cough and shortness of breath in the patients with ILD and the relatively small number of patients in our study, these findings should be interpreted with caution.
The present study also found that the disease duration was shorter in the fibrosing ILD group than in the nonfibrosing ILD group. This finding may be related to the more severe nature of the lung injury and the more rapid disease progression. Moreover, the presence of dry cough, shortness of breath, and fever may also encourage the patients to seek medical attention, thus shortening the course of the disease. Therefore, early detection of pulmonary fibrosis in patients with pSS is essential. A study from Norway showed that patients with mixed connective tissue–associated severe pulmonary fibrosis had a shorter mean duration of the disease than patients with mild to moderate fibrosis.24 In addition, the patients with the most severe pulmonary fibrosis showed faster disease progression than those with a less severe condition.24
In our study, the frequency of positive anti‑SSA and anti‑Ro52 antibodies was significantly lower in the fibrosing ILD group than in the nonfibrosing ILD group. ANAs have been suggested as a risk factor for pulmonary involvement in patients with SS.9 Moreover, another study concluded that anti‑Ro52, anti‑SSB, and anti‑Ro60 antibodies and ANA positivity rates are not significantly different in patients with pSS and with or without ILD.25 Other studies have shown that patients with pSS and pulmonary involvement have a significantly lower rate of anti‑SSA antibody positivity than controls.8 These differences may be due to discrepancies in the study populations, regions, inclusion criteria, and laboratory testing methods.
As a noninvasive test, high‑resolution CT (HRCT) can provide a sensitive diagnosis of lung lesions at the early stages.26 The most common HRCT manifestation of pSS‑ILD is nonspecific interstitial pneumonia (NSIP); other manifestations include organizing pneumonia and usual interstitial pneumonia (UIP).27 According to Park et al,28 patients with NSIP and patients with UIP plus collagen vascular disease have similar prognoses. Similarly, the prognoses of patients with pSS‑UIP and pSS‑NSIP do not differ and are only influenced by the severity of reticular formation observed on HRCT.29 Moreover, pulmonary fibrosis is observed not only in UIP types but also in other types of ILD, such as fibrous NSIP.15 Therefore, in this study, we focused on the HRCT signs of pSS‑ILD rather than on classifying patients with simple ILD by lung involvement.
The distribution of chest CT abnormalities in our study was not uniform. The most common chest CT abnormality was ground‑glass opacity, a finding confirmed by previous studies.7,11,30,31 Ground‑glass opacity can be attributed to exudation and inflammation of the lung parenchyma.32 However, when combined with traction bronchiectasis and / or reticular abnormalities, it may also be a part of the fibrotic process.15 This explains why the frequency of ground‑glass opacity was significantly higher in the fibrosing ILD group than in the nonfibrosing ILD group in our study. First, the ground‑glass opacity representing the fibrotic component (when coexisting with reticular abnormalities and / or traction bronchodilation) was significantly more predominant in the fibrosing ILD group. Second, owing to pulmonary fibrosis, pulmonary function is impaired and more likely to be combined with pulmonary inflammation, resulting in increased ground‑glass opacity.
In our study, we also found that interlobular septal thickening, consolidation shadows, subpleural lines, bronchial wall thickening, bronchiectasis, pleural thickening, and mediastinal lymph node enlargement occurred significantly more frequently in patients with pSS‑associated pulmonary fibrosis, which may indicate that these patients had more severe chest CT abnormalities and were more likely to have mediastinal lymph nodes and pleural lesions. Additionally, pSS also commonly manifests in the mediastinum as lymphadenopathic and lymphoplasmacytic inflammation.33 It is well known that pleurisy is most commonly found in patients with systemic lupus erythematosus and that pleural involvement in pSS is relatively uncommon.34,35 A previous study reported that patients with UIP or pSS commonly showed bronchial wall thickening and mediastinal lymphadenopathy on chest CT and more pronounced pathological changes, such as bronchiolitis, cysts, plasma cell infiltration, and pleurisy.36 These results are similar to those observed in the present study.
We found changes on chest CT images in 69 of 151 patients with pSS‑ILD, most of whom were treated with immunosuppressive drugs and glucocorticoids. Overall, the absorption, lack of CT changes, and progression did not differ significantly in the 2 groups. However, the rate of progression was higher, and the rate of focal resorption was lower in the patients with pSS‑associated pulmonary fibrosis than in those with pSS but without fibrosis. This may indicate that glucocorticoids effectively reduce the inflammatory response in the lungs.37 Furthermore, patients with pSS and pulmonary fibrosis have an unfavorable response to immunosuppressive drugs and a worse prognosis than patients with pSS without fibrosis.38
Our study had some limitations. First, it was a retrospective work with relatively few cases, which may have influenced the results. Second, some relevant laboratory test results were missing. Future studies with a strict, prospective design are required to further analyze the relationship between laboratory data and pSS‑related pulmonary fibrosis. Third, only 32 of the 151 patients underwent pulmonary function tests, and many indices of pulmonary function were missing, which prevented us from conducting an in‑depth study of the relationship between CT and pulmonary function in the patients with pSS‑ILD. Finally, supporting pathology data were lacking in this study. However, all relevant pathological examinations are invasive, making it difficult for most patients to provide consent.
In conclusion, this study demonstrated that pSS‑associated pulmonary fibrosis might have a short course and be associated with various CT manifestations, including lobular septal increase, ground‑glass opacity, pleural lesions, and mediastinal lymph node enlargement. Dry cough and shortness of breath are potential predictors of pulmonary fibrosis in patients with pSS. Assessing both the chest CT presentation and clinical symptoms can help evaluate the patients with PSS‑ILD and provide a basis for clinical intervention.
- Mariette X, Criswell LA. Primary Sjögren’s syndrome. N Engl J Med. 2018; 378: 931‑939. | Crossref
- Chung A, Wilgus ML, Fishbein G, Lynch JP. Pulmonary and bronchiolar involvement in Sjögren’s syndrome. Semin Respir Crit Care Med. 2019; 40: 235‑254. | Crossref
- Gupta S, Ferrada MA, Hasni SA. Pulmonary manifestations of primary Sjögren’s syndrome: underlying immunological mechanisms, clinical presentation, and management. Front Immunol. 2019; 10: 1327. | Crossref
- Antoniou KM, Margaritopoulos GA, Tomassetti S, et al. Interstitial lung disease. Eur Respir Rev. 2014; 23: 40‑54. | Crossref
- Lonzetti L, Zanon M, Pacini GS, et al. Magnetic resonance imaging of interstitial lung diseases: a state‑of‑the‑art review. Respir Med. 2019; 155: 79‑85. | Crossref
SUPPLEMENTARY MATERIAL
ARTICLE INFORMATION