Allogeneic hematopoietic stem cell transplantation for acquired severe aplastic anemia: a summary of a 20-year experience
Key words: allogeneic hematopoietic stem cell transplantation, immunosuppressive treatment, infections, outcome, severe aplastic anemia



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Allogeneic hematopoietic stem cell transplantation for acquired severe aplastic anemia: a summary of a 20-year experience
Introduction: Severe aplastic anemia (SAA) is a rare but potentially fatal disorder characterized by hypocellular bone marrow and resulting in pancytopenia. It can be cured with allogeneic hematopoietic stem cell transplantation (allo‑HSCT), especially in young individuals.
Objectives: The main objective of the study was to assess the safety of the procedure and to identify the factors influencing long‑term post‑transplant outcome.
Patients and methods: Using our institutional database, we performed a retrospective analysis of the patients with SAA allotransplanted in the years 2001–2021.
Results: Seventy patients (49 men) at a median age of 25 years at transplantation underwent allo‑HSCT. Thirty‑eight patients received immunosuppressive treatment (IST) before transplantation. Twenty‑one patients received grafts from human leukocyte antigen–matched sibling, 44 from unrelated donors, and 5 from haploidentical related donors. Peripheral blood remained the source of stem cells in the majority of patients. Primary graft failure was observed in 2 cases. The incidence of acute graft‑versus‑host disease (GVHD) was 44%, whereas chronic GVHD was observed only in 4 patients. Median follow‑up was 3 years (interquartile range, 0.45–11.5). Post‑transplant outcome was comparable between patients with upfront allo‑HSCT and those who relapsed after IST. In the univariable analysis, only a higher Eastern Cooperative Oncology Group (ECOG) score at transplantation and infections in the post‑transplant period were found to be associated with unfavorable outcome. Fifty‑three patients were alive at last contact. Most transplanted patients died due to infectious complications. A 2‑year overall survival was 73%.
Conclusions: The results of allo‑HSCT in SAA are satisfactory and offer long‑term survival and good quality of life. Higher ECOG score and the presence of infections are associated with poor post‑transplant outcome.
What's new?
Allogeneic hematopoietic stem cell transplantation provides excellent outcomes in young individuals with severe aplastic anemia, but it may be questionable in patients older than 40 years. Among 70 allotransplantations performed in our center for the last 20 years, 80% of the procedures were done in patients up to the age of 40 years. The estimated overall survival exceeded 70% at 2 years. The use of treosulfan‑based conditioning resulted in a low incidence of graft failure and severe acute graft‑versus‑host disease. There was no difference in post‑transplant outcomes between peripheral blood and bone marrow stem cell transplantations. The use of an alternative donor was associated with an increased risk of death. Infections were common and remained the main cause of death. Nowadays, new possibilities emerged even for older patients and for those lacking a human leukocyte antigen–matched donor. Our data suggest that allogeneic transplantation may be an option not only for young patients, and alternative donor search should be performed in selected cases.
Introduction
Aplastic anemia (AA) is a rare but potentially fatal disorder characterized by hypocellular bone marrow and pancytopenia in the peripheral blood. AA may be accompanied by paroxysmal nocturnal hemoglobinuria (PNH) clone. The severity of AA is determined by neutrophil count. The treatment of severe aplastic anemia (SAA) with absolute neutrophil count (ANC) below 0.5 × 109/l has significantly improved in recent years. Allogeneic hematopoietic stem cell transplantation (allo‑HSCT) remains the treatment of choice, especially in young patients below 40 years old with identical sibling donor (ISD).1,2
The widely approved guidelines recommend a course of immunosuppressive treatment (IST) containing antithymocyte globulin (ATG) and cyclosporine with an addition of thrombopoietin agonist eltrombopag, before a patient with SAA receives a transplant from a non‑ISD.3 In older patients, treatment decisions are based not only on the disease‑related factors but also on their overall condition and comorbidities, so as to reduce the risk of treatment‑related toxicity that may contribute to high morbidity and mortality rate.4 In the case of older patients and / or those who lack an ISD, it is recommended to proceed with IST as the first‑line treatment.1 Of note is that approximately one‑third of the patients do not respond to the IST and one‑third may relapse after the primary response. A clonal evolution may be observed in some patients.5,6
Due to the high failure rate of IST, in relapsed or refractory (r/r) cases, human leukocyte antigen (HLA)-matched unrelated allo‑HSCT remains an option. Nowadays, also haploidentical donor (HID) transplantation is an attractive option in patients with r/r SAA.7 Upfront HID transplantation remains a solution for young patients who lack a sibling or an unrelated donor (URD). Another important issue is the availability of HID and a short time from the donor identification to the collection of the stem cells.
Here, we report the results of our retrospective analysis of 70 allo‑HSCTs for SAA performed in our center in adult patients over the last 20 years.
Patients and methods
For the retrospective analysis, the medical records of the SAA patients who underwent allo‑HSCT between January 2001 and December 2021 were identified in our institutional database. The diagnosis of acquired SAA was based on the Camitta criteria,8 which include hemoglobin concentration below 100 g/l, platelet count below 50 × 109/l, and ANC below 1.5 × 109/l. The modified Camitta criteria8,9 were used to assess the disease severity. Our transplant policy was that all SAA patients below 40 years of age are qualified for upfront ISD transplantation following a careful assessment of comorbidities. In the absence of an HLA‑identical sibling donor, an unrelated donor transplant procedure was planned. For patients aged 40 years and older, IST remains the first‑line treatment. The patients who failed to respond to the IST were qualified for transplant procedure if their Hematopoietic Cell Transplant Comorbidity Index was low (total score ≤2). Alternative donors, such as haploidentical ones, were considered only in selected patients who failed to respond to the IST and / or if other matched donors (ISD or URD) were unavailable.
The patients were screened for PNH using flow cytometry on peripheral blood to detect deficiency of glycophosphatidyl‑inositol anchored proteins, such as CD14, CD16, and CD24, as well as fluorescent aerolysin for white blood cells, and CD55 and CD59 for red cell analysis. The patients with coexisting subclinical PNH clone size below 30% detected on flow cytometry were included in this analysis. None of these patients presented hemolysis or other PNH symptoms. Definitions of engraftment and graft failure were consistent with the previously published criteria.10 Transfusions of irradiated leukodepleted blood products and other supportive care measures were performed as per institution practices. Standard antibiotic, antifungal, and antiviral prophylaxis was applied in all cases. The patients received standard Pneumocystis jiroveci prophylaxis for at least 6 months after the transplantation or longer in the individuals with prolonged immunosuppressive treatment due to chronic graft‑versus‑host disease (GVHD).
Day‑30 donor chimerism in unsorted bone marrow cells was assessed after the transplantation using the short tandem repeats method, and was calculated following the defined genetic profiles of the donor and the recipient. The patients were monitored for cytomegalovirus (CMV) reactivation by weekly measurement of CMV copy number by polymerase chain reaction in the serum until day +100 or longer, depending on the need for prolonged immunosuppressive therapy. Acute and chronic GVHD (aGVHD and cGVHD, respectively) were diagnosed and graded according to standard criteria.11 Due to the retrospective design of the study some data were incomplete or unavailable.
Statistical model building and evaluation
The Mann–Whitney test was applied to compare the median age of the transplanted patients between the first (2001–2010) and the second decade (2011–2021).
Due to the specific type of patient survival data, in which fatal events were recorded only in the first 2 years of the follow‑up (and a distribution of the follow‑up times, which was skewed and excessively asymmetric), we first fitted different statistical models and validated them using the Akaike information criterion (AIC) (data not shown).12 The distribution of overall survival (OS) was estimated using the Royston–Parmar proportional hazards approach13 with a flexible baseline (https://cran.r‑project.org/web/packages/flexsurv/vignettes/flexsurv‑examples.pdf). The number of knots was chosen to minimize AIC (in our flexible fully‑parametric alternative we implemented a spline with 1 internal knot). Based on the collected dataset, hazard ratios with 95% CIs and P values below 0.1 were reported, whereas P values below 0.05 were considered significant. The computation was performed using the R statistical platform.14
Results
Patient characteristics
Seventy patients (49 [70%] men and 21 [30%] women) with SAA at a median age of 25 years at transplant (interquartile range [IQR], 20–34) underwent allo‑HSCT between January 2001 and December 2021. Thirty‑eight patients (54%) received IST before transplantation. The median time from diagnosis to transplant was 6 months (IQR, 4–12) for the whole group of patients, 4 months (IQR, 4–12) in the case of the upfront procedures, and 9.5 months (IQR, 6–17) for the r/r cohort. All patients were transfusion‑dependent at the time of the transplant. Cytogenetics was available only for 16 patients (23%), and the majority of the tested patients (62.5%) presented with normal diploid karyotype, whereas single chromosome abnormalities were detected in 2 cases (12.5%), and unsuccessful cytogenetics was found in 4 patients (25%). In the remaining cases the results were not available. Liver function impairment up to 6 months before the diagnosis of SAA was noted in 10 patients (14%). Hepatitis that would fulfil the criteria for hepatitis‑associated aplastic anemia (HAAA) was documented in 1 case only. The PNH clone was found in 6 out of 41 tested patients (15%). The majority of patients (79%) had good performance status, that is, Eastern Cooperative Oncology Group (ECOG) score 0 or 1 at transplant. Nonetheless, nearly half of the patients had a history of a chronic disease or a serious infection prior to the transplant procedure. Descriptive characteristics of the patients are provided in Table 1.
Parameter | Value | |
Data are presented as number (percentage) or median (IQR).
Abbreviations: ATG, antithymocyte globulin; HAAA, hepatitis‑associated aplastic anemia; IQR, interquartile range; IST, immunosuppressive therapy; PNH, paroxysmal nocturnal hemoglobinuria; UC, unsuccessful cytogenetics; r/r, relapsed / refractory | ||
Age, y | 25 (20–34) | |
Men / women | 49 (70)/21 (30) | |
Karyotype | Normal | 10 (14) |
Abnormal | 2 (3) | |
UC | 4 (6) | |
No data available | 54 (77) | |
Subclinical PNH clone | 6 (9) | |
Prior ATG‑based IST | 38 (54) | |
Prior HAAA | 1 (1.5) | |
Transfusion dependency | 70 (100) | |
Interval from diagnosis to transplantation, mo | All patients | 6 (4–12) |
Upfront group | 4 (2–6.5) | |
r/r group | 9.5 (6–17) | |
Transplant data
Median donor age was 28 years (IQR, 23.5–38.5), and 70% (49/70) of the donors were men. As many as 21 patients (30%) received their graft from an ISD, and an HLA‑matched unrelated donor (MUD) was found in 31 cases (44%). There was an HLA mismatch in 13 cases (19%) (3 cases of allelic mismatch and 10 cases of antigen mismatch). A HID was used in 5 transplant procedures (7%). Eligible HIDs included related family members who shared at least 1 HLA haplotype. Thirty‑one patients (44%) presented with unfavorable CMV serological status in donor‑recipient pairs (D/R) before the transplant procedure (3 cases of D+/R–, 28 cases of D+/R+). In 4 cases (6%), the status was favorable (D–/R–), and in the remaining 32 cases (46%) the status was intermediate (D–/R+) in terms of CMV reactivation risk after transplantation. In 3 cases (4%), the data were missing. Twenty‑eight patients (40%) presented with AB0 group‑matched donors. The major AB0 group mismatch was detected in 14 cases (20%), in 19 cases (27%) it was minor, and in 8 D/R pairs (11%) major and minor AB0 group incompatibilities (bidirectional mismatch) were present. No data were available in 1 case (2%). Stem cell source was granulocyte‑colony stimulating factor (G‑CSF) mobilized peripheral blood in 47 cases (67%), and bone marrow in 20 cases (29%). Both sources of stem cells were used in 3 procedures. In general, bone marrow grafts had a median CD34+ cell count of 2.31 × 106/kg of the recipient body weight (IQR, 1.7–3.23), whereas the peripheral stem cell grafts of 6.41 × 106/kg body weight of the recipient (IQR, 4.52–7.72). For the whole group of patients, median CD34+ cell count was 5.64 × 106/kg of the recipient body weight (IQR, 3.07–7.08). The predominant conditioning regimen was based on treosulfan and cyclophosphamide in 44 cases (63%). A total of 18 patients (26%) received conditioning with cyclophosphamide alone. We performed 5 haploidentical procedures (7%), with standard Baltimore protocol containing fludarabine, cyclophosphamide, and total body irradiation (TBI) 200 cGy with post‑transplant cyclophosphamide. In the remaining 3 procedures (4%), nonmyeloablative conditioning regimens were applied in 2 cases (fludarabine- or TBI‑based), and a myeloablative regimen (busulfan- and cyclophosphamide‑based) was used in 1 case only.
To reduce the risk of cGVHD, all patients received in vivo T‑cell depletion with rabbit ATG. Sixty‑three out of 70 patients (90%) received cyclosporine containing GVHD prophylaxis and a short course of methotrexate. Only 1 patient (1.5%) received mycophenolate mofetil with a short course of methotrexate. In 1 case (1.5%), tacrolimus and methotrexate were administered. In 5 cases of haploidentical grafts (7%), the immunosuppressive treatment contained a standard dose of post‑transplant cyclophosphamide, and mycophenolate mofetil and tacrolimus starting from day +5.
There were no significant differences in engraftment in patients receiving G‑CSF stimulated peripheral blood and bone marrow grafts (18 days for neutrophils; IQR, 14–21 and 16 days for platelets; IQR, 12–21). Post‑transplant G‑CSF was given at 5 µg/kg to 10 patients out of 70 (14%). Thirteen patients remained transfusion‑dependent at discharge. In those cases tapering of standard IST, low doses methylprednisolone and / or eltrombopag were needed to stimulate engraftment. The transplant data are presented in Table 2.
Parameter | Value | |
Data are presented as number (percentage) or median (IQR).
Abbreviations: Bu, busulfan; CMV, cytomegalovirus; CMV serostatus (D+/R–), serological status of CMV in donor / recipient pairs; CSA, cyclosporine; Cy, cyclophosphamide; ECOG, Eastern Cooperative Oncology Group; Flu, fludarabine; ISD, identical sibling donor; GVHD, graft‑versus‑host disease; MMF, mycophenolate mofetil; MMUD, mismatched unrelated donor; MTX, methotrexate; MUD, matched unrelated donor; TAC, tacrolimus; TBI, total body irradiation; Treo, treosulfan; Tx, transplant; others, see Table 1 | ||
ECOG score (pre‑transplant) 0–1 / ≥2 | 55 (79)/15 (21) | |
Recipient age, y | 25 (20–34) | |
Recipients aged ≤ 40 y | 56 (80) | |
Donor age, y | 28 (23.5–38.5) | |
Female donor to male recipient | 14 (20) | |
ISD / MUD / MMUD / haploidentical | 21 (30) / 31 (44) / 13 (19) / 5 (8) | |
CMV serostatus (D+/R– or D+/R+) | 31 (44) | |
AB0 matched grafts | 28 (40) | |
Stem cell source | Bone marrow | 20 (29) |
Peripheral blood | 47 (67) | |
Both | 3 (4) | |
Conditioning regimen | Treo/Cy/ATG | 44 (63) |
Cy/ATG | 18 (26) | |
Flu/Cy/TBI | 5 (7) | |
Flu/Cy/ATG | 2 (3) | |
Bu/Cy/ATG | 1 (1) | |
GVHD prophylaxis | CSA/MTX | 63 (90) |
TAC/MTX | 1 (1.5) | |
MTX/MMF | 1 (1.5) | |
post‑Tx Cy, TAC, MMF | 5 (7) | |
Number of transplanted CD34+ cells, × 106/kg body weight | All grafts | 5.64 (3.07–7.08) |
Bone marrow grafts | 2.31 (1.7–3.23) | |
Peripheral stem cells grafts | 6.41 (4.52–7.72) | |
Number of transplanted CD3+ cells × 107/kg | 21.05 (11.55–30.8) | |
Days to neutrophil engraftment | 18 (14–21) | |
Days to platelet engraftment | 16 (12–21) | |
Day‑30 donor chimerism | 100 (97–100) | |
Survival outcome
Median follow‑up was 3 years (IQR, 0.45–11.5). In general, 53 patients (76%) were alive at last contact, transfusion‑independent, and without any evidence of clonal evolution or secondary malignancy. As many as 17 patients (24%) died. Six patients (9%) died before the recovery of hematopoietic system due to enteritis caused by Clostridioides difficile and concurrent sinusoidal obstruction syndrome (n = 1), meningitis (n = 1), pneumonia (n = 2), sudden cardiac death during sleep in a patient with no prior cardiovascular history (n = 1), and multiorgan failure due to toxicity of the conditioning regimen (n = 1).
Data on day‑30 donor chimerism in unsorted bone marrow cells were available for 73% of the patients. Median percentage of donor chimerism in the study group was 100% (IQR, 97%–100%). Forty patients (57%) developed infectious complications until day +30 after the transplantation. Seventeen patients (24%) were diagnosed with bacterial bloodstream infections caused by Klebsiella pneumoniae (n = 7), Enterobacter cloacae (n = 3), Enterococcus faecalis (n = 2), Escherichia coli (n = 2), Staphylococcus hemolyticus (n = 1), Staphylococcus epidermidis (n = 1), and Pseudomonas aeruginosa (n = 1). Serious infections included pneumonia caused by Klebsiella pneumoniae ESBL (n = 5), Aspergillus spp. (n = 4), Candida spp. (n = 3), Pneumocystis jiroveci (n = 5), massive fungal sinusitis (n = 2), infection at the site of the central vein catheter (n = 5), enteritis due to Clostridioides difficile infection (n = 2), and meningitis (n = 2). Viral complications included hemorrhagic cystitis caused by BK virus (n = 8) and CMV reactivation (n = 4).
The incidence of primary graft failure was 3% (2 patients). Both patients were retransplanted. One of them engrafted and is still alive. The second one died from acute grade 4 GVHD. Secondary graft failure among patients with initial engraftment was observed in 1 patient, who was successfully retransplanted and is still alive. The incidence of aGVHD in patients who engrafted was 44% (28 patients), and 3 patients developed grade 3–4 GVHD, and eventually died due to steroid refractoriness. cGVHD was observed in 4 patients (6%), but none of them presented with extensive cGVHD. All outcome data are presented in Table 3.
Parameter | Value | |
Data are presented as number (percentage) or median (IQR).
| ||
Primary graft failure | 2 (3) | |
Secondary graft failure | 1 (1.5) | |
Acute GVHD | 28 (44) | |
Chronic GVHD | 4 (6) | |
Infectious complications by day +30 | 40 (57) | |
Deceased | Before day +30 | 6 (9) |
After day +30 | 11(16) | |
Alive at last contact | 53 (76) | |
Follow‑up, y | 3 (0.45–11.5) | |
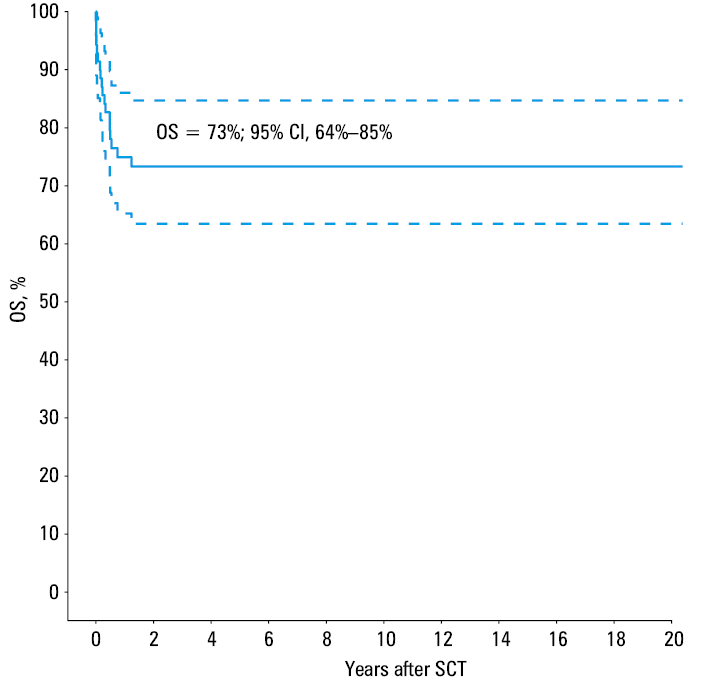
Two‑year OS rate was 73% (Figure 1). There was no statistical difference in the post‑transplant outcome between the upfront and IST patients. In univariable analysis, only the ECOG score at transplant and infections in post‑transplant period were found to be associated with unfavorable outcome. Higher ECOG score (≥2) resulted in a nearly 2‑fold increase in the risk of death (P = 0.02). Deterioration of performance status on admission was associated with coexisting infections in 14 cases (20%), whereas in 1 case the patient was diagnosed with femoral neck fracture due to femoral head necrosis in the course of steroid treatment. Infections diagnosed in the post‑transplant period were found to correlate with inferior outcome (P = 0.04). In our study group, 17 patients (24%) died due to severe infectious complications (in 3 cases associated with grade 4 aGVHD). We observed 6 early deaths (9%), while 11 patients (16%) died later with a median of 122 days after the transplantation (from 51 days to 14 months).
Transplantation from a nonsibling donor (unrelated or alternative) increased the death risk more than twice. No significant improvement in survival among patients transplanted in the years 2001–2010 (n = 37) and 2011–2021 (n = 33) was demonstrated. An analysis of the second decade showed that the risk of death was almost 2.5 times higher than in the first decade. There was a significant (P <0.001) and almost 10‑year difference in the median age of patients transplanted in the first and the second decade, namely 23 and 32 years, respectively. Univariable analysis of the risk factors is presented in Table 4.
Discussion
SAA is a rare, life‑threatening bone marrow failure usually diagnosed in the setting of peripheral pancytopenia and hypocellular bone marrow. In approximately 5% of patients, SAA can be preceded by elevated transaminases and hyperbilirubinemia, which may be associated with a viral etiology.15 It is a heterogenous disease that may be either inherited or acquired. Inherited bone marrow failure syndromes are rare genetic diseases, typically diagnosed in children and associated with an increased risk of coexisting malignancy.16 Most patients diagnosed with SAA are sporadic immune‑mediated cases. The pathophysiology of acquired SAA is unknown, although it is believed that a dysregulated immune response leads to autoreactive T‑cell destruction of hematopoietic stem and progenitor cells in a genetically susceptible patient.17 Another factor that may contribute to the development of SAA may be exposure to some toxic substances or viral infection, but usually the triggering antigen remains unknown. In some cases, SAA may be associated with cytogenetic abnormalities, most often trisomy.18-20 Nearly half of the patients with acquired SAA may develop subclinical PNH clone.21
In approximately 1% to 5% of patients, AA features may be preceded by hepatitis, that is, HAAA.22 It takes usually up to 6 months until pancytopenia occurs. The etiology is unknown.22 In our study group, there was 1 patient diagnosed with HAAA.
The incidence of acquired SAA is approximately 2 per 1 000 000 people per year in Europe and North America, and it occurs mostly in young adolescents (aged 15–25 years) and in elderly patients over 60 years of age. The men‑to‑women ratio is approximately 1:1. This incidence is 2 to 3 times higher in Asia, which may be explained by some genetic or environmental factors.15 In our study group, the majority of the patients were men, their median age at the time of transplant was 25 years, and 26% of the patients were below the age of 20 years.
Our report provided some interesting and novel findings. The use of treosulfan‑based conditioning (63% of the transplanted patients) seems to translate into low incidence of severe aGVHD (4%) and cGVHD (6%). Moreover, the incidence of primary graft failure was only 3%. Our results seem to be much better than those presented by others.23 It was earlier demonstrated that the use of peripheral blood as a source of stem cells was associated with worse outcome.24 However, no difference in post‑transplant outcome between grafts from peripheral blood and bone marrow was found in our study. It was also reported previously that patients who received grafts from unrelated donors fared worse than those transplanted from matched siblings,24 and this was confirmed in our study. Infections were claimed to be the main cause of post‑transplant death,25,26 and actually 17 patients from our study died from these complications. OS at 2 years was 73%, and it was lower than that reported by the European Group for Blood and Marrow Transplantation (EBMT) (89%).27 However, one should bear in mind that our transplanted patients were older than those in the EBMT report (25 years vs 20 years), and age remains a very strong predictor of the post‑transplant outcome.1,28
Patients with SAA require early diagnosis and intervention, because the time between diagnosis and treatment is believed to be one of the predictors of survival, as the patient may acquire HLA‑antibodies due to frequent transfusion need. There is also a significant risk of developing serious infections, especially bacterial and fungal ones due to prolonged neutropenia.1 In our patient group, the median time from diagnosis to transplantation was different for the upfront and r/r cohort, but it did not impact the survival.
According to the literature, the prognosis for SAA with supportive care only is dismal, with mortality rates exceeding 80% at 2 years.15 Immunosuppressive therapy combines ATG and cyclosporine in most cases. OS rate at 2 years after IST with cyclosporine and ATG is approximately 60%–65%.5 Attempts to add G‑CSF not only did not improve the response rates, but were found to increase the risk of clonal evolution.27,29 It was also proved that horse ATG was superior to rabbit ATG.5 The response rates were significantly improved by adding eltrombopag, an oral thrombopoietin receptor agonist, to standard IST, without exacerbating side effects.3,30 Contrary to allo‑HSCT, immunosuppressive therapy causes less severe adverse events, but is associated with significantly greater risk of relapse and exposes the patient to long‑term immunosuppression with a possibility of developing clonal hematopoiesis or even cytogenetic abnormalities.31 The 2 major complications of allo‑HSCT in SAA are graft failure and GVHD. GVHD, which may be observed even in 50% of patients, provides no beneficial effect in AA patients, and its risk should be reduced as much as possible. Furthermore, cGVHD is one of the factors that can shorten survival. Allo‑HSCT is associated with greater toxicity, not only early toxicity due to the conditioning regimen, but also late one, such as hormonal imbalance.1
Steady improvement was postulated24in the outcomes of patients who were offered transplantation over the past years, especially in terms of unrelated donor transplantations, which are noninferior to sibling transplantations. We compared the outcomes of transplantations performed in our center in the years 2001–2010 and 2011–2021, but we did not notice any improvement in survival. This may be partly explained by the fact that the patients transplanted before 2011 were younger (median age, 22 years; IQR, 17–52) than those transplanted from 2011 onward (median age, 30 years; IQR, 18–62). Age was proven to be a strong predictor in sibling transplants, indicating that patients aged below 20 years have an 86% chance of a 10‑year survival, those aged 21–40 years a 76% chance, whereas in individuals over 40 the odds are only 55%.28 Unfortunately, the survival in the latest patient group has not improved significantly over the last years.32 For unrelated donors, the chances of a 5‑year survival are 77% for patients aged 11–30 years, 66% for patients aged 30–40 years, and only 49% for individuals aged above 40 years.1 Another issue is the source of stem cells. In our study, we performed 19 bone marrow transplantations in the first decade (in 3 cases additional collection of stem cells from peripheral blood was needed) and 18 mobilized peripheral blood stem cell transplantations. In the second decade, the majority of grafts were collected from stimulated peripheral blood (bone marrow was the source of stem cells in only 3 patients), which may explain stable transplant results in our center. The use of peripheral blood stem cell grafts is high (even up to 60%), which was noted in the EBMT analysis of transplants in SAA.24 In that study, marrow grafts correlated with significantly superior survival rate (5‑year OS, 80% vs 70%) when compared with peripheral stem cell grafts.24
The standard conditioning regimen for ISD under the age of 40 years is cyclophosphamide 200 mg/kg body weight and ATG. Fludarabine‑based conditioning appears to be a good option especially in elderly patients due to reduced toxicity.33,34 In our study group, fludarabine‑based conditioning regimens were applied in the minority of patients, whereas the majority received treosulfan‑based conditioning, and we did not observe any graft failure in this group. Treosulfan as a prodrug and a structural analogue of busulfan shows myeloablative, immunosuppressive, and antimalignancy activity, as well as low organ toxicity, which was proven in many studies, mostly in leukemia patients,35 but also in SAA36 and other nonmalignant diseases.37
To sum up, many authors agree that bone marrow transplantation after ATG‑based conditioning regimen with cyclophosphamide and GVHD prophylaxis with cyclosporine and methotrexate provides successful engraftment and excellent survival rate especially in younger individuals.1,38,39 In our study group, all patients (apart from haploidentical procedures) received in vivo T‑cell depletion with rabbit ATG. We observed low incidence of grade 3–4 aGVHD (4%). Nonetheless, 44% of the patients presented mild symptoms of grade 1–2 aGVHD. Chronic limited GVHD was observed only in 4 patients (6%), and none of the patients developed extensive symptoms.
In recent years, HLA‑haploidentical transplantation using post‑transplant cyclophosphamide (PT‑Cy) conditioning regimens proved to be a reasonable option for patients needing a rapid transplant procedure or for those who do not have an HLA‑matched donor or who failed IST. Post‑transplant PT‑Cy has greatly improved the safety and efficacy of alternative donor bone marrow transplantation (BMT) by facilitating engraftment and decreasing the risk of GVHD. Due to higher graft failure rates and increased cGVHD risk, HID transplantation should be considered a salvage option. The risk of rejection justifies adding a small dose of TBI. The data published by the EBMT concerning 33 HID transplantations in Europe in the years 2011–2017, revealed 2‑year OS of 78%, with low incidence of aGVHD (23% until day +100) and cGVHD (10% at 2 years).7 A most recent study40 indicated that haploidentical procedures should be implemented in clinical practice as a salvage treatment in SAA patients. The most important issue is to obtain high stem cell dose (>2.5 × 108 nucleated marrow cells per kg body weight) from bone marrow harvests.40 The reported 1‑year survival was 81%, and it is important to highlight that the study group included children.40 Our experience in the field of HID transplantations has so far been limited, but in our group 2 out of 5 patients are alive. Median recipient age at transplant in that group was 33 years (IQR, 28–45). Three patients died due to serious infections, aGVHD, or graft failure.
Management of AA patients with infection remains a challenge. Infections have been regarded by some authors as an adverse factor associated with unfavorable transplantation outcome.34,41 However, other data did not confirm a negative impact of an active infection on survival in SAA patients,25 indicating that transplantation may provide relatively fast neutrophil recovery, minimalize development of invasive fungal infections and therefore increase the chances for long‑term survival. Some data demonstrated that cGVHD and transplant‑related mortality rates were significantly higher in the group of SAA patients who presented with infection.26 Our data indicated that coexisting infection was a factor significantly related to dismal prognosis after the transplant procedure.
If we compare SAA patients with those suffering from other hematologic malignancies, including leukemia, the latter include a heavily pretreated population frequently colonized by many pathogens and often presenting with a history of organ dysfunction and bloodstream infection. It can be also the case in SAA, however, the interval between diagnosis and transplantation is usually shorter for SAA, especially when a sibling donor is available. Besides, SAA patients do not receive chemotherapy prior to transplantation. If we consider post‑transplant end points, graft failure risk is lower in leukemia patients but the incidence of GVHD, including severe one, is significantly higher in leukemia than in SAA. The rate of infections seems to be similar.42
Poor performance score was found to be a significant factor limiting survival of SAA patients following BMT from alternative donors,43 which is consistent with our findings.
The data from the EBMT analysis24 of 1448 SAA transplantations indicate that the strongest negative predictor of survival is the use of stimulated peripheral blood grafts (in both sibling and unrelated transplantations). The other prognostic factors include the interval from diagnosis to transplant, age, CMV donor / recipient serostatus, and stem cell source. The latest data also confirm that bone marrow should be the preferred stem cell source for MUD transplantation in ATG‑based conditioning regimens.2 Some authors state that the donors willing to donate bone marrow stem cells should be favored over those willing to give peripheral stem cells.24
A small proportion of our transplanted patients presented with subclinical PNH clone; however, all these patients met the SAA criteria. None of them received eculizumab, a monoclonal antibody against C5 protein before transplantation. The outcome of patients with and without subclinical PNH clone was comparable (data not shown).
Conclusions
The results of allo‑HSCT in SAA are satisfactory and promise long‑term survival and good quality of life. Higher ECOG score and the presence of infections are associated with poor post‑transplant outcome.
- Bacigalupo A. How I treat acquired aplastic anemia. Blood. 2017; 129: 1428‑1436. | Crossref
- Snowden JA, Sánchez‑Ortega I, Corbacioglu S, et al; European Society for Blood and Marrow Transplantation (EBMT). Indications for haematopoietic cell transplantation for haematological diseases, solid tumours and immune disorders: current practice in Europe, 2022. Bone Marrow Transplant. 2022; 19: 1‑23. | Crossref
- Peffault de Latour R, Kulasekararaj A, Iacobelli S, et al; Severe Aplastic Anemia Working Party of the European Society for Blood and Marrow Transplantation. Eltrombopag added to immunosuppression in severe aplastic anemia. N Engl J Med. 2022; 386: 11‑23.
- Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006; 108: 2509‑2519. | Crossref
- Scheinberg P, Nunez O, Weinstein B, et al. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med. 2011; 365: 430‑438. | Crossref
ARTICLE INFORMATION