Pulmonary proteinosis and secondary hemophagocytic syndrome in a patient with lysinuric protein intolerance



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Pulmonary proteinosis and secondary hemophagocytic syndrome in a patient with lysinuric protein intolerance
Lysinuric protein intolerance (LPI) is a rare, autosomal recessively inherited metabolic disease, caused by a mutation within the gene SCL7A7 on chromosome 14q11. The disease is characterized by intolerance of protein‑rich foods and secondary disruption of the urea cycle. The highest incidence of LPI has been observed in Finland (1 per 60 000 births), Italy, and Japan. There are no available epidemiologic data for Poland.1
The presented patient had protein intolerance with severe gastrointestinal symptoms and hyperammonemia since birth. Genetic testing performed at the age of 4 years revealed a compound heterozygote of c(572_573delAA);(446_447insC) in the SLC7A7 gene.
A 19‑year‑old woman was admitted to the hospital in a very severe condition with symptoms of respiratory failure (SpO2 35 mm Hg). Additional tests showed an elevated C‑reactive protein level of 25 mg/dl (reference range [RR], 0–0.8 mg/dl), elevated white blood count (WBC) of 16 × 109/l (RR, 4–10 × 109/l), normocytic and normochromic anemia with hemoglobin of 10 g/dl (RR, 11.1–14.7 g/dl), lactate dehydrogenase (LDH) of 250 U/l (RR, 135–223 U/l), and transferrin of 152 mg/dl (RR, 200–360 mg/dl). Chest computed tomography (CT) showed extensive interstitial and parenchymal lesions with reduced volume of the lower lobes of the lungs (Figure 1A). Empirical antibiotic therapy (ceftrioxone, ciprofloxacin), steroid therapy (methylprednisolone 250 mg/day), and oxygen therapy through a mask (12 l/min) were started. Diagnostics for infectious agents included blood (negative for Epstein–Barr virus, cytomegalovirus, and HIV), urine (negative for Legionella pneumophila and Streptococcus pneumoniae antigens), bronchial aspirate (negative for Mycobacterium tuberculosis, Pneumocystis, and galactomannan), and a nasal swab (negative for SARS‑CoV‑2 and influenza). Analysis of the bronchial aspirate showed acidophilic granular masses (Periodic acid–Schiff [PAS]+) and macrophages with foamy cytoplasm (PAS+). Pulmonary proteinosis was diagnosed and the patient was qualified for whole lung lavage (WLL). Under general anesthesia, the patient was intubated with a double lumen endotracheal tube. Under gravity, 800‑ml portions of heated 0.9% NaCl (total of 10 l) were infused into the isolated lung. Subsequently, mechanical drainage of the chest was performed using a cough assist device, and the fluid was removed under gravity. The steps were repeated until a clear fluid was obtained.2,3
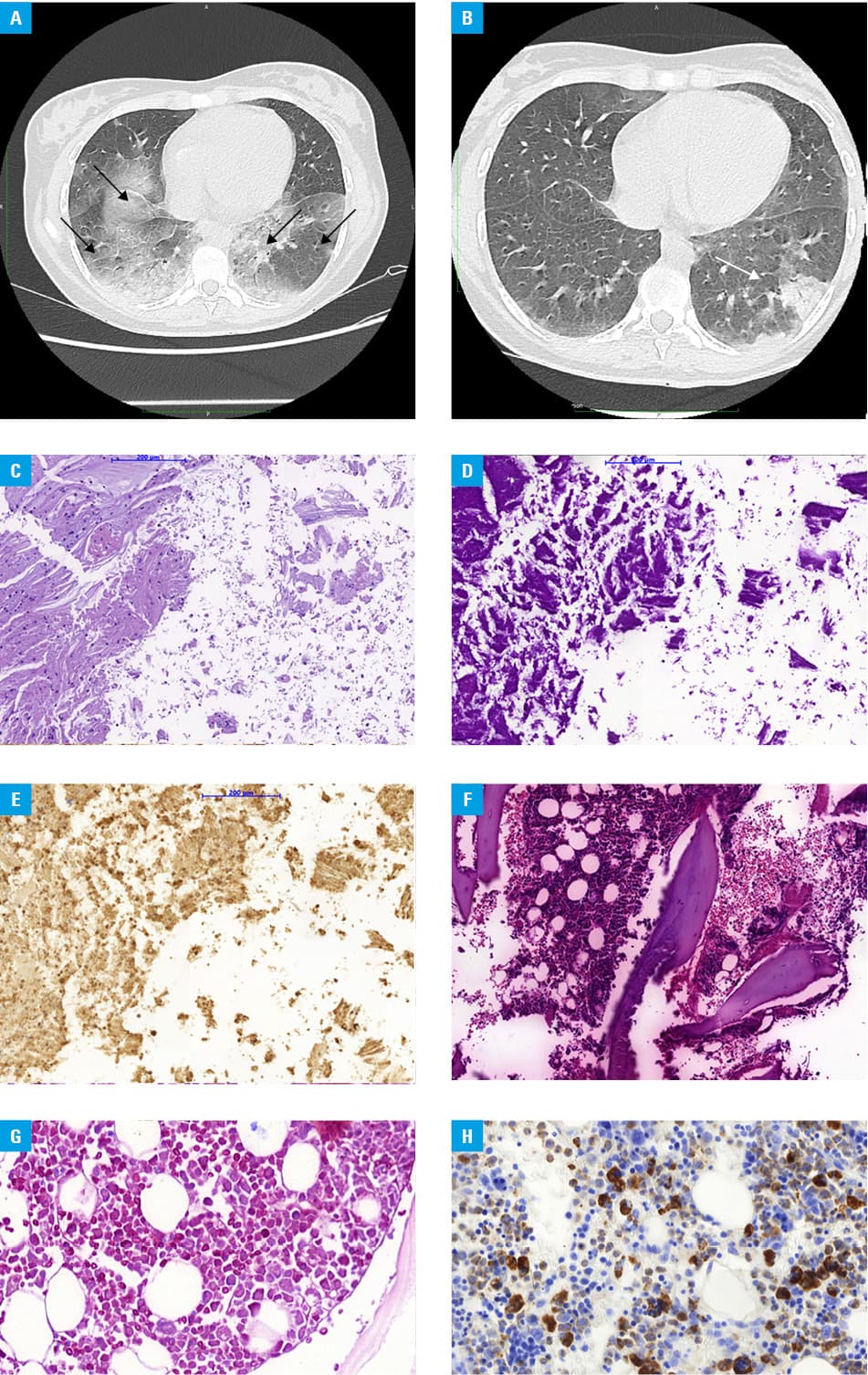
After 2 days, the second lung lavage procedure was performed. After another 2 days, respiratory failure resolved with SpCO2 38.6 mm Hg and SpO2 80.8 mm Hg.
After 18 months, the patient was readmitted to the hospital due to a fever of 38 °C. In additional tests infectious factors were excluded. Laboratory workup showed decreased WBC of 2.98 × 109/l, anemia with hemoglobin level of 7.9 g/dl, iron level of 52 µg/dl (RR, 60–180 µg/dl), total iron binding capacity of 181 U/l (RR, 228–428 U/l), ferritin 21 937 µg/ml (RR, 13–150 µg/ml), LDH 2486 U/I; and triglycerides of 253 mg/dl (RR, 35–165 mg/dl).
A chest CT scan showed extensive foci of ground‑glass opacity and reduction of lesions present on previous scans (Figure 1B).
Bone marrow trepanobiopsy revealed rich‑cell smears with macrophages (CD68KP1+, CD68PGM1+) phagocytosing nucleated cells, granulocytic lineage (MPO+) with predominantly immature forms, and red cell lineage (CD71+) with slight dyskeratosis (Figure 1C–1H). Karyotype examination from the bone marrow showed no clonality. Secondary hemophagocytic syndrome (HLH) was diagnosed.
Initial treatment involved dexamethasone 8 mg/m2 and human normal immunoglobulin 0.5 g/kg body weight. Due to the treatment failure after 3 months, rituximab 375 mg/m2 every 4 weeks was added. After 4 months of treatment, cyclosporine 200 mg/day was started.
The patient has maintained satisfactory respiratory status for the next 32 months. For the last 6 months, there have been no symptoms of generalized inflammatory response syndrome, which were assessed during control visits.
In the treatment of interstitial lung disease due to LPI, WLL is more effective than steroids.3 The occurrence of HLH is a severe complication, difficult to treat and significantly increasing the risk of death.4,5
- Palacín M, Bertran J, Chillarón J, et al. Lysinuric protein intolerance: mechanisms of pathophysiology. Mol Genet Metab. 2004; 81: S27‑S37. | Crossref
- Awab A, Khan MS, Youness HA. Whole lung lavage‑technical details, challenges and management of complications. J Thorac Dis. 2017; 9: 1697‑1706. | Crossref
- Santamaria F, Montella S, Mirra V, et al. Respiratory manifestations in patients with inherited metabolic diseases. Eur Respir Rev. 2013; 22: 437‑453. | Crossref
- Malinowska I, Machaczka M, Popko K, et al. Hemophagocytic syndrome in children and adults. Arch Immunol Ther Exp (Warsz). 2014; 62: 385‑394. | Crossref
- Matsukawa Y, Sakamoto K, Ikeda Y, et al. Familial hemophagocytic lymphohistiocytosis syndrome due to lysinuric protein intolerance: a patient with a novel compound heterozygous pathogenic variant in SLC7A7. Int J Hematol. 2022; 116: 635‑638. | Crossref
ARTICLE INFORMATION