From Cushing syndrome to lipodystrophy: an ultrarare case of MFN2-associated lipomatosis



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
From Cushing syndrome to lipodystrophy: an ultrarare case of MFN2-associated lipomatosis
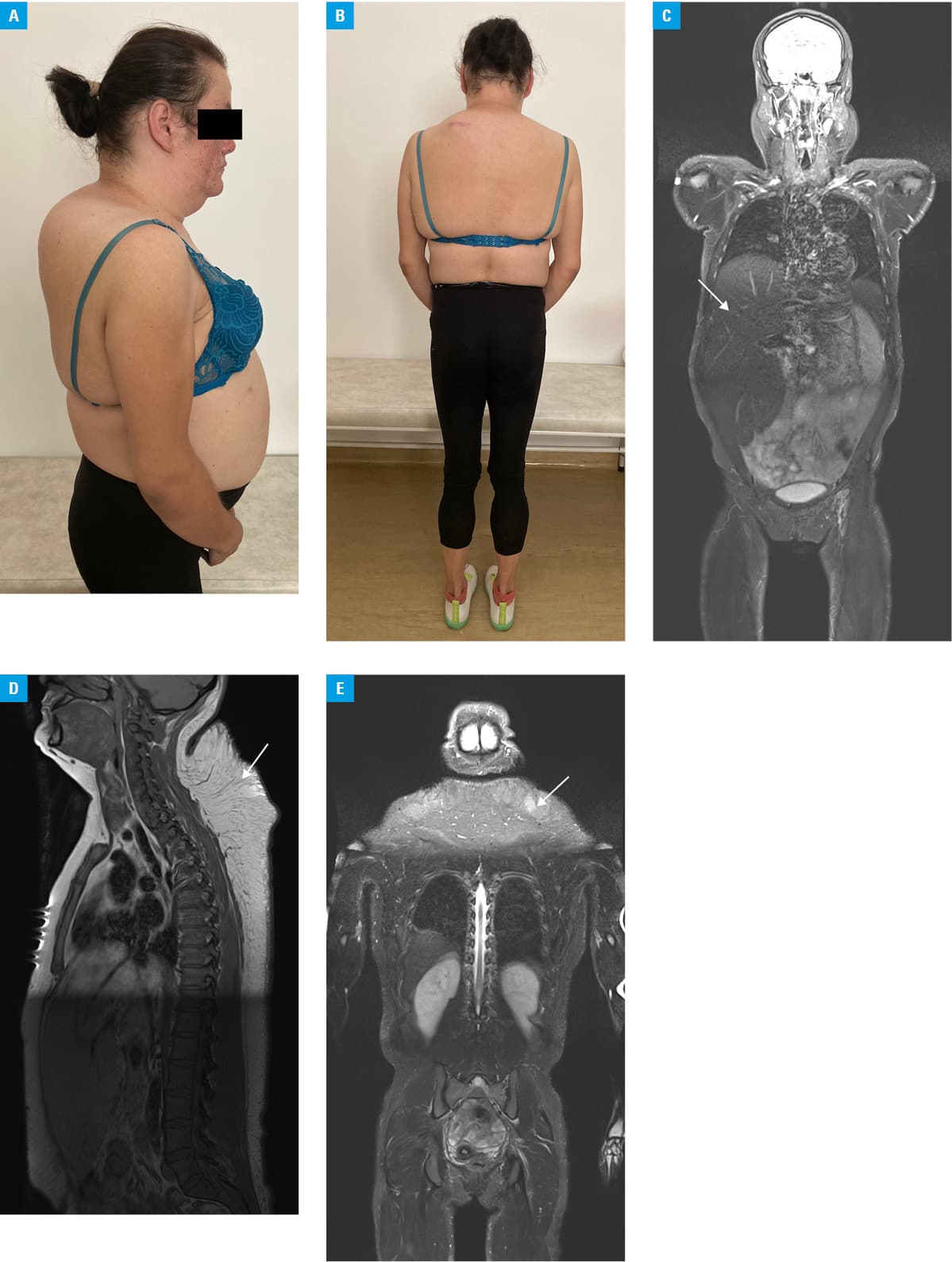
A 33‑year‑old woman was admitted to a hospital due to abdominal pain and severe hypertriglyceridemia. The patient was also diagnosed with type 2 diabetes, hypertension, high insulin resistance, and polycystic ovary syndrome. She was hospitalized twice for Cushingoid appearance (Figure 1A and 1B) that appeared in her early twenties, but based on laboratory and imaging tests, the Cushing syndrome was ruled out. Laboratory workup revealed mild hypertransaminasemia, severe hypertriglyceridemia, high ferritin levels, increased levels of lactic acid, and very low leptin levels (0.33 ng/l; reference range, 3.7–11.1 ng/l). Hemochromatosis, Wilson disease, and autoimmune hepatitis were excluded. Magnetic resonance imaging (MRI) revealed fatty liver, hepatomegaly (Figure 1C), excessive accumulation of subcutaneous fat in the neck and back, as well as almost complete disappearance of subcutaneous fat in the arms, hips, and legs (Figure 1C–1E). In addition, MRI of the left hip joint showed left femoral head of reduced height with subsequent increased coverage by the acetabulum. Diffuse reduction of fat content in the bone marrow of bone elements of unclear etiology has also been described. At the age of 34, the patient began to report paresthesia in the feet and an increasing pain in the extremities of a polyneuropathic nature. Electromyographic examination confirmed low‑grade demyelinating sensory polyneuropathy and moderate axonal‑demyelinating motor polyneuropathy with significant axonal loss in the right peroneal nerve and slow conduction in all distal nerves of the lower limbs. Due to suspected partial lipodystrophy, the patient was referred to a genetic clinic. Genetic testing did not confirm the presence of a mutation in the LMNA gene, mutations of which are responsible for the majority of lipodystrophies with familial partial lipodystrophy phenotype. Subsequently, the whole exome sequencing was performed. The pathogenic c.2119C>T p.(Arg707Trp) variant (frequency of occurrence in the population described in databases of 0.0251%) and a novel c.1496‑2A>G variant (frequency of 0.0003977%) were found, each in a single copy of the mitofusin‑2 (MFN2) gene. We performed a segregation analysis for the presence and independent inheritance of the 2 identified altered MFN2 alleles with the Sanger sequencing. The result of the segregation analysis was consistent with autosomal recessive inheritance. It showed that the unaffected mother was a carrier of the variant c.2119C>T, whereas the novel c.1496‑2A>G variant was inherited from the unaffected father. The novel substitution, c.1496‑2A>G, is located at the 3’ end of intron 14, at a canonical acceptor site, which was confirmed by the in silico analysis. The predictions with Human Splice Finder and NetGene2 (v2.42) indicated that the substitution removes the acceptor splice site. The in silico analysis with the use of Combined Annotation Dependent Depletion tool (https://cadd.gs.washington.edu/snv, Accessed March 27, 2023), (score 34) and Functional Analysis through Hidden Markov Models (v2.3) (https://fathmm.biocompute.org.uk/fathmmMKL.htm, Accessed March 27, 2023), (score 0.96) indicated that this splicing variant is deleterious.
The Online Mendelian Inheritance in Man database did not link the mutations in the MFN2 gene with lipodystrophy. Mutations in the MFN2 gene have previously been shown to be responsible for the development of a peripheral neuropathy called the Charcot‑Marie‑Tooth disease.
In recent years, there have been reports describing patients with a homozygous p.(Arg707Trp) variant in the MFN2 gene presenting with multiple symmetric lipomatosis with neuropathy.1,2 Then, it was revealed that also biallelic MFN2 mutations and at least 1 p.(Arg707Trp) allele induce mitochondrial dysfunction leading to upper body adipose hyperplasia and suppression of leptin expression.3
In conclusion, only a review of the available literature made it possible to diagnose our patient with MFN2-associated lipomatosis, classified as lipodystrophy, and caused by biallelic mutations of the MFN2 gene, one of which is the p.(Arg707Trp) variant. To the best of our knowledge, this case is only the third report describing a biallelic mutation in this gene.
Lipodystrophies are rare diseases characterized by a total or partial loss of adipose tissue.4 Lack of subcutaneous depots and ectopic fat tissue accumulations in the liver, muscle, pancreas, and blood vessels lead to metabolic disorders, such as insulin resistance, diabetes mellitus, nonalcoholic fatty liver disease, and hypertriglyceridemia. Treatment involves intensive management of metabolic complications. Results from studies using a leptin analogue for treatment are promising, however, the drug is not widely available due to its cost and registration limitations.5
The rare occurrence of lipodystrophy, a clinical picture similar to the Cushing syndrome, and difficulties in reaching the diagnosis based on genetic testing may result in underdiagnosis. Increased awareness of the disease may not only contribute to a more frequent diagnosis of lipodystrophy, but also to interdisciplinary efforts to improve the therapy and the availability of already existing treatment with leptin analogues.
- Sawyer SL, Ng ACH, Innes AM, et al. Homozygous mutations in MFN2 cause multiple symmetric lipomatosis associated with neuropathy. Hum Mol Genet. 2015; 24: 5109‑5114. | Crossref
- Capel E, Vatier C, Cervera P, et al. MFN2-associated lipomatosis: clinical spectrum and impact on adipose tissue. J Clin Lipidol. 2018; 12: 1420‑1435. | Crossref
- Rocha N, Bulger DA, Frontini A, et al. Human biallelic MFN2 mutations induce mitochondrial dysfunction, upper body adipose hyperplasia, and suppression of leptin expression. Elife. 2017; 6: e23813. | Crossref
- Brown RJ, Araujo‑Vilar D, Cheung PT, et al. The diagnosis and management of lipodystrophy syndromes: a multi‑society practice guideline. J Clin Endocrinol Metab. 2016; 101: 4500‑4511. | Crossref
- Araujo‑Vilar D, Santini F. Diagnosis and treatment of lipodystrophy: a step‑by‑step approach. J Endocrinol Invest. 2019; 42: 61‑73. | Crossref
ARTICLE INFORMATION