Diffuse alveolar hemorrhage in a chronically hemodialyzed patient with progressive necrotic skin lesions as a manifestation of calciphylaxis



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Diffuse alveolar hemorrhage in a chronically hemodialyzed patient with progressive necrotic skin lesions as a manifestation of calciphylaxis
Calciphylaxis is the ectopic calcification in the medial layer of small and medium‑caliber arteries that predominantly affects the dermis and subcutaneous tissue.1 It is a complication in up to 4% of the end‑stage kidney disease (ESKD) population, especially of those treated with maintenance hemodialysis (HD), and occurs sporadically in nonuremic individuals. The risk factors include female sex, diabetes mellitus, autoimmune disorders, obesity, hyperphosphatemia, use of calcium‑based phosphate binders, active vitamin D metabolites, and the use of corticosteroids and vitamin K antagonists.1,2
The main symptom of calciphylaxis are skin lesions that develop in several stages: nodules, livedo reticularis and livedo racemosa, hemorrhagic bullae, ulcerations, and necrosis.2 The lesions tend to appear on otherwise healthy skin subjected to a minor trauma, and occur at adipose‑rich sites, in proximal areas of the lower extremities, the abdomen, but also distally. Histopathology shows circumferential calcium deposits in the medial wall of small arterioles, with signs of ischemic necrosis in the dermis and subcutaneous tissue, as well as endovascular fibrosis and intimal proliferation.1
Strong pain often resistant to traditional analgesics is a hallmark of calciphylaxis lesions; most patients require complex pain management, often with opioid agents.
Establishing diagnosis is challenging, as there are no specifically designed tests, imaging methods, or biomarkers. Calciphylaxis is often diagnosed by exclusion of other likely pathologies;2 sometimes it becomes apparent after unsuccessful treatment of other conditions. Differential diagnosis must include cellulitis, infection, peripheral artery disease, vasculitis, antiphospholipid syndrome, or warfarin‑induced skin necrosis. Radiological testing, that is, X‑ray, computed tomography (CT), or magnetic resonance imaging may reveal areas of calcification, which is helpful but not required in the diagnosis of calciphylaxis.1 The most effective diagnostic method remains deep punch biopsy of the skin and subcutaneous tissue, with cross‑sections of small vessels and optional staining for calcium deposits. However, as calciphylaxis causes ischemia of the tissue, the biopsy entails a high risk of bleeding, complicated wound healing, and potential infection of the affected site.
Currently there are no guidelines or approved treatments for the condition.3 The key treatment objectives are optimized dialysis, correction of calcium‑phosphate (Ca‑P) disturbances, including secondary or tertiary hyperparathyroidism (HPT), as well as proper wound care and pain management. Most regimes include longer and more frequent HD sessions with low‑calcium dialysate solutions, and a conversion to hemodiafiltration (HDF) may be considered. To improve Ca‑P balance, most patients are switched to noncalcium based phosphate binders, such as sevelamer.3 The classic chelating agent used in calciphylaxis, sodium thiosulfate (STS), aims to remove deposits of calcium apatite from the vessels. Other pharmacotherapies with unproven effectiveness include bisphosphonates and vitamin K2. Due to difficulties in the diagnosis and lack of effective treatment, the 1‑year mortality of this complication ranges from 45% to 80%, and proximal lesions are associated with worse survival, mainly due to wound infections.1
A 49‑year‑old, slim woman with ESKD due to membranoproliferative glomerulonephritis, chronically hemodialyzed for 9 years was admitted to a nephrology ward with severe leg pain lasting for 4 to 6 weeks, rated as 7/10, exacerbated during HD sessions. She described the pain as a burning, splitting sensation, localized in both calves and the frontal surface of the thighs.
Her comorbidities included Crohn disease in remission for over 2 years, severe HPT treated with cinacalcet, hypertension, myocardial infarction in 2016, and paroxysmal atrial fibrillation treated with vitamin K antagonists (VKAs) for 5 months.
Physical examination revealed palpable, firm subcutaneous tumors on both thighs and calves, with areas of livedo reticularis on both lower limbs (Figure 1A). Other abnormalities included symmetrical, reduced vesicular sound over upper lung fields. The laboratory tests showed anemia (hemoglobin 7.0 g/dl; reference range [RR], 12–16 g/dl), elevated C‑reactive protein (38 mg/l; RR, <5.0 mg/l), with normal level of procalcitonin, hyperphosphatemia (2.49 mmol/l; RR, 0.81–1.45 mmol/l), hypocalcemia (2.09 mmol/l; RR, 2.20–2.65 mmol/l), and elevated parathormone (1135 pg/ml; RR, 10–60 pg/ml).
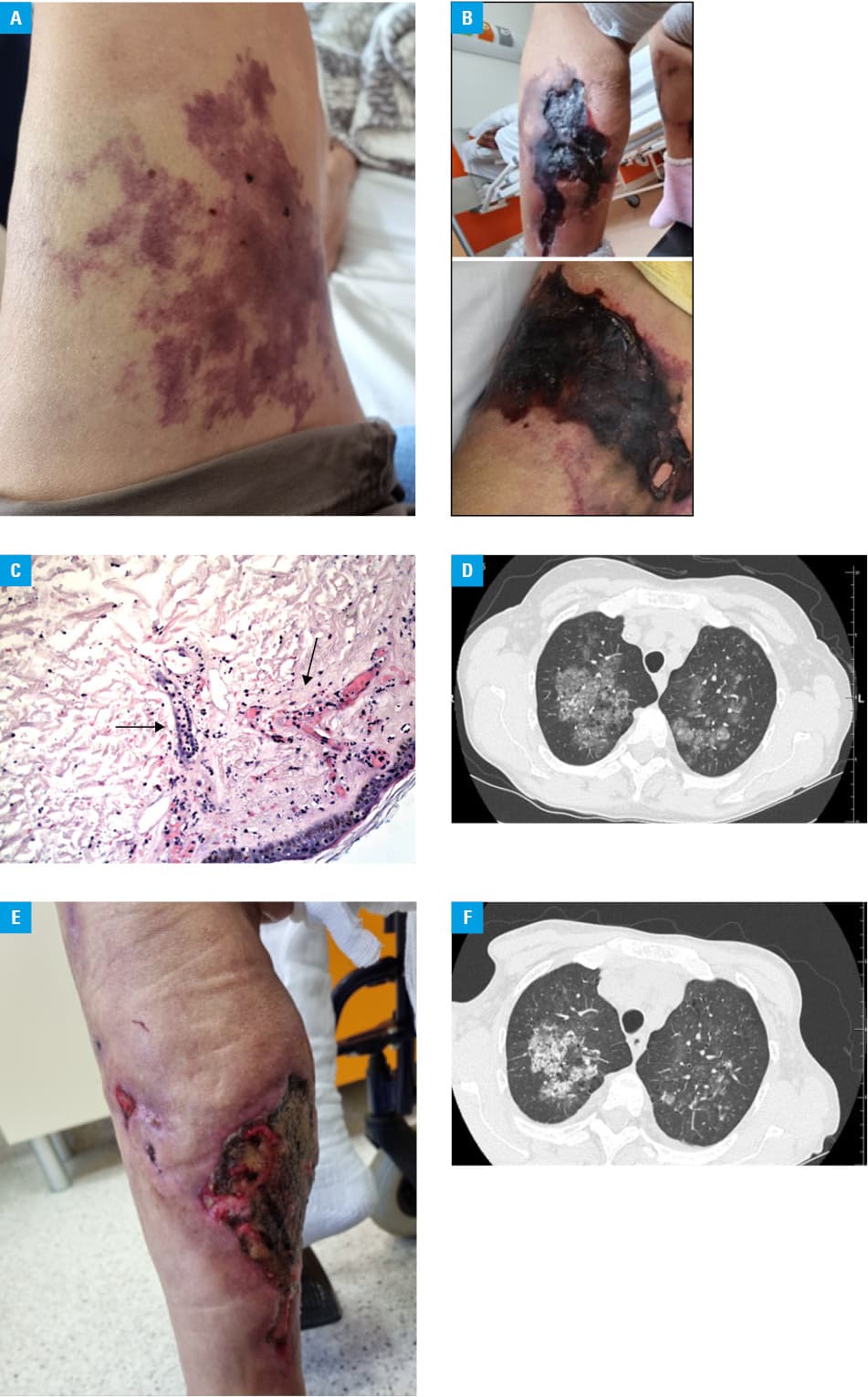
Due to a suspicion of erythema nodosum, typical in patients with Crohn disease, systemic steroids (glucocorticoids [GCSs]), that is, methylprednisolone, followed by oral prednisone, were introduced. In the following days the patient showed no clinical response, and her general condition deteriorated. She complained of progressive exacerbated pain and muscle weakness in the lower extremities. Her gait was severely impaired and ultimately rendered her unable to walk; skin lesions progressed into areas of livedo racemosa and necrosis (Figure 1B). The Guillain–Barre syndrome and myasthenia gravis were excluded. Laboratory tests revealed increased serum levels of creatinine kinase (1138 U/l; RR, 0–145 U/l), as well as increased activity of transaminases: aspartate aminotransferase (57 U/l; RR, 0–35 U/l) and alanine transaminase (58 U/l; RR, 0–35 U/l), and normal bilirubin levels. Erythema nodosum was excluded, and further differential diagnoses involved arterial / venous ischemia, steroid‑induced myopathy, and dermatomyositis. Doppler ultrasound excluded arterial occlusion and venous thrombosis. Excisional skin biopsy revealed leukocytoclastic vasculitis (Figure 1C), with the absence of antinuclear antibodies and antineutrophil cytoplasmic antibodies in the serum. The high‑resolution computed tomography (HRCT) of the lungs revealed diffuse alveolar hemorrhage (DAH) in both lungs (Figure 1D), confirmed by bronchoalveolar lavage, surprisingly without hemoptysis or dyspnea. At that time, upper gastrointestinal bleeding was diagnosed with a Forrest III lesion on gastroscopy, with exacerbation of anemia requiring blood cell transfusion. Since clinical data strongly indicated seronegative vasculitis with DAH, immunosuppressive therapy with intravenous cyclophosphamide (2 pulses of 600 mg), followed by rituximab (1 pulse of 600 mg) was administered. For the following 2 weeks, the patient showed no clinical response, the skin lesions progressed over new areas, with necrosis in both proximal and distal areas of the legs (Figure 1B), and new areas of livedo racemosa on the left arm, which later also progressed into ulcerations. Due to the pain that was particularly strong during HD sessions, and forced the patient to end them earlier, high doses of opioid analgesics were introduced.
Considering the overall clinical presentation with progressive skin necrosis located mainly in the proximal extremities and concomitant DAH, no response to immunosuppression, long history of HPT and VKA treatment, severe calciphylaxis with pulmonary involvement was diagnosed. Immunosuppression was discontinued, HD was intensified with conversion to HDF with simultaneous infusions of STS during the last 30 minutes of each dialysis session. Cinacalcet was continued, and calcium‑based phosphate binders were replaced with sevelamer. Anticoagulant treatment with low‑molecular‑weight heparin was continued. Hyperbaric oxygen therapy was included to support wound healing, and the patient was under surgical supervision. During a 5‑month follow‑up the patient general status improved, she started a physical therapy, the lesions showed areas of demarcation, with margins of healthy tissue around them (Figure 1E). CT showed reduced pulmonary involvement (Figure 1F).
The presented case provided great diagnostic difficulties. In most cases, a diagnosis of calciphylaxis is that of exclusion, and the reported misdiagnosis rate is over 70%.1 Our patient presented many risk factors for calciphylaxis, such as a long history of HPT with deep Ca‑P disturbances, previous use of VKAs and GCSs, autoimmune conditions, such as membranoproliferative glomerulonephritis, and Crohn disease.1,2 The last 2 conditions potentially suggested an immunological nature of the symptoms. The initial differential diagnoses relied mainly on skin lesions and laboratory findings. Moreover, histopathologic reports mentioned no classic features of calciphylaxis, such as calcium deposits and necrosis,2 instead suggesting vasculitis. This prompted us to perform HRCT, which revealed DAH. DAH is a potentially fatal manifestation of vasculitis, and that combined with the rapidly deteriorating clinical status of the patient necessitated remission‑inducing treatment. Other causes of DAH, such as connective tissue disorders, antiphospholipid syndrome, and infections were excluded. Cyclophosphamide, at a dose adjusted for kidney function, was administered according to the guidelines, followed by a pulse of rituximab. Neither medication yielded any results, the general condition of the patient, including her mental wellbeing, deteriorated. The necrotic lesions progressed to new areas. The lack of response to immunosuppression increased the likelihood of calciphylaxis, thus the search for available medical literature on the cases of solid organ calciphylaxis commenced. There are only a few cases of DAH due to calciphylaxis described in the literature with available imaging (complete CT scans or cross‑sections) to compare with the patient’s results.4,5 Following pulmonological and radiological consultations, the diagnosis of calciphylaxis was confirmed and appropriate treatment was implemented.3 After 5 months of unspecific, multimodal treatment of calciphylaxis the patient’s general condition improved, lung lesions are reduced, and vitamin K2 at 1 mg a day orally was added to the treatment.
Calciphylaxis is a rare but severe complication of ESKD. Calcium deposits accumulate slowly and covertly, clinical manifestations become apparent only at advanced stages. Skin lesions are the main clinical sign of calciphylaxis; however, the disease may affect solid organs and mimic other conditions, including vasculitis. The key to diagnosing calciphylaxis is including this complication in differential diagnosis in the patients with risk factors, mainly with severe Ca‑P imbalance during maintenance HD. The diagnosis relies on exclusion of other possible conditions, as it has no specific laboratory or radiological markers, while the disease itself carries a high risk of complications and death.
- Gallo Marin B, Aghagoli G, Hu SL, et al. Calciphylaxis and kidney disease: a review. Am J Kidney Dis. 2023; 81: 232‑239. | Crossref
- Baby D, Upadhyay M, Joseph MD, et al. Calciphylaxis and its diagnosis: a review. J Family Med Prim Care. 2019; 8: 2763. | Crossref
- Zoi V, Bacharaki D, Sardeli A, et al. Calciphylaxis: a long road to cure with a multidisciplinary and multimodal approach. Case Rep Nephrol. 2022; 2022: 3818980. | Crossref
- Dhanjal TS, Babu SB, Beevers G, Ferner RE. Calciphylaxis associated with widespread pulmonary calcification. BMJ Case Rep. 2009; 2009: bcr09.2008.0880. | Crossref
- Matsuo T, Tsukamoto Y, Tamura M, et al. Case report acute respiratory failure due to “pulmonary calciphylaxis” in a maintenance haemodialysis patient. Nephron. 2001; 87: 75‑79. | Crossref
ARTICLE INFORMATION