Genetic predisposition to differentiated thyroid cancer in the Polish population
1 ,
Key words: genetics, Polish population, thyroid cancer, whole genome sequencing
,
Key words: genetics, Polish population, thyroid cancer, whole genome sequencing

 CC BY 4.0
CC BY 4.0
Genetic predisposition to differentiated thyroid cancer in the Polish population
Introduction: Genome sequencing technologies reveal molecular mechanisms of differentiated thyroid cancer (DTC). Unlike somatic mutation analysis from thyroidectomy samples, germline mutations showing genetic susceptibility to DTC are less understood.
Objectives: The study aimed to assess the prevalence of germline mutations predisposing to DTC in a cohort of Polish individuals based on their whole genome sequencing data.
Patients and methods: We analyzed sequencing data from 1076 unrelated individuals totaling over 1018 billion read pairs and yielding an average 35.26 × read depth per genome, released openly for academic and clinical research as the Thousand Polish Genomes database (https://1000polishgenomes.com). The list of genes chosen for further analysis was based on the review of previous studies.
Results: The cohort contained 104 variants located within the coding and noncoding DNA sequences of 90 genes selected by ClinVar classification as pathogenic and potentially pathogenic. The frequency of variants in the Polish cohort was compared with the frequency estimated for the non‑Finnish European population obtained from the gnomAD database (gnomad.broadinstitute.org). Significant differences in variant frequency were found for the APC, ARSB, ATM, BRCA1, CHEK2, DICER1, GPD1L, INSR, KCNJ10, MYH9, PALB2, PLCB1, PLEKHG5, PTEN, RET, SEC23B, SERPINA1, SLC26A4, SMAD3, STK11, TERT, TOE1, and WRN genes.
Conclusions: Even though the Polish population is genetically similar to the other European populations, there are significant differences in variant frequencies contributing to the disease development and progression, such as those in the RET, CHEK2, BRCA1, SLC26A4, or TERT genes. Further studies are needed to identify genomic variants associated directly with DTC.
What's new?
Despite progress in elucidating molecular mechanisms underlying differentiated thyroid cancer (DTC) thanks to genome sequencing, germline mutations responsible for genetic susceptibility to DTC are still poorly recognized. Since the genetic background predisposing to DTC may differ among populations, the aim of this study was to assess the prevalence of genetic germline mutations predisposing to DTC in a cohort of Polish individuals, using the whole genome analysis. To date, this is the first analysis harnessing whole genome sequencing data for the Slavic population, with Slavs accounting for over 4.5% of the world’s inhabitants. The frequency of variants discovered in the Polish cohort, a genetic reference for the Slavic population, was compared with the variant frequency estimated for the non‑Finnish European population obtained from the gnomAD database. The results showed significant variant frequency differences in the APC, ARSB, ATM, BRCA1, CHEK2, DICER1, GPD1L, INSR, KCNJ10, MYH9, PALB2, PLCB1, PLEKHG5, PTEN, RET, SEC23B, SERPINA1, SLC26A4, SMAD3, STK11, TERT, TOE1, and WRN genes.
Introduction
Thyroid cancer (TC) is the most common malignant endocrine tumor.1 It accounts for 1%–2% of all neoplasms.2 Its prevalence is steadily and the most rapidly rising among all cancers.3 This increasing trend may be partly linked to an improved detection of smaller (<2 cm) tumors due to more frequent and better ultrasound evaluation, fine‑needle aspiration biopsy, and increased pathologic reporting of incidental microcarcinomas.4 In 2020, the World Health Organization (Global Cancer Statistics 2020: GLOBOCAN) reported 586 202 new cases of TC, of which 43 646 resulted in death.5
TC types are classified according to their histologic characteristics (Figure 1).6-10 Differentiated TC (DTC) is also known as nonmedullary TC (NMTC), and represents approximately 90% of all TCs.11 Over 90% of TCs are sporadic, due to somatic genetic alterations.12 Only 3%–9% of all TCs are familial nonmedullary TC (FNMTC) cases, defined by the presence of TC in 2 or more first‑degree relatives, in the absence of predisposing environmental factors.13 FNMTC can be divided into syndromic or nonsyndromic FNMTC. It depends on whether the cancer is a part of one of many constellations of tumors (syndromic) or a primary cancer (nonsyndromic FNMTC).14 Out of all FNMTCs, only 5% of the syndromic forms have well‑defined driver germline mutations. On the contrary, 95% of FNMTCs are nonsyndromic with less well‑defined genetic susceptibility.14
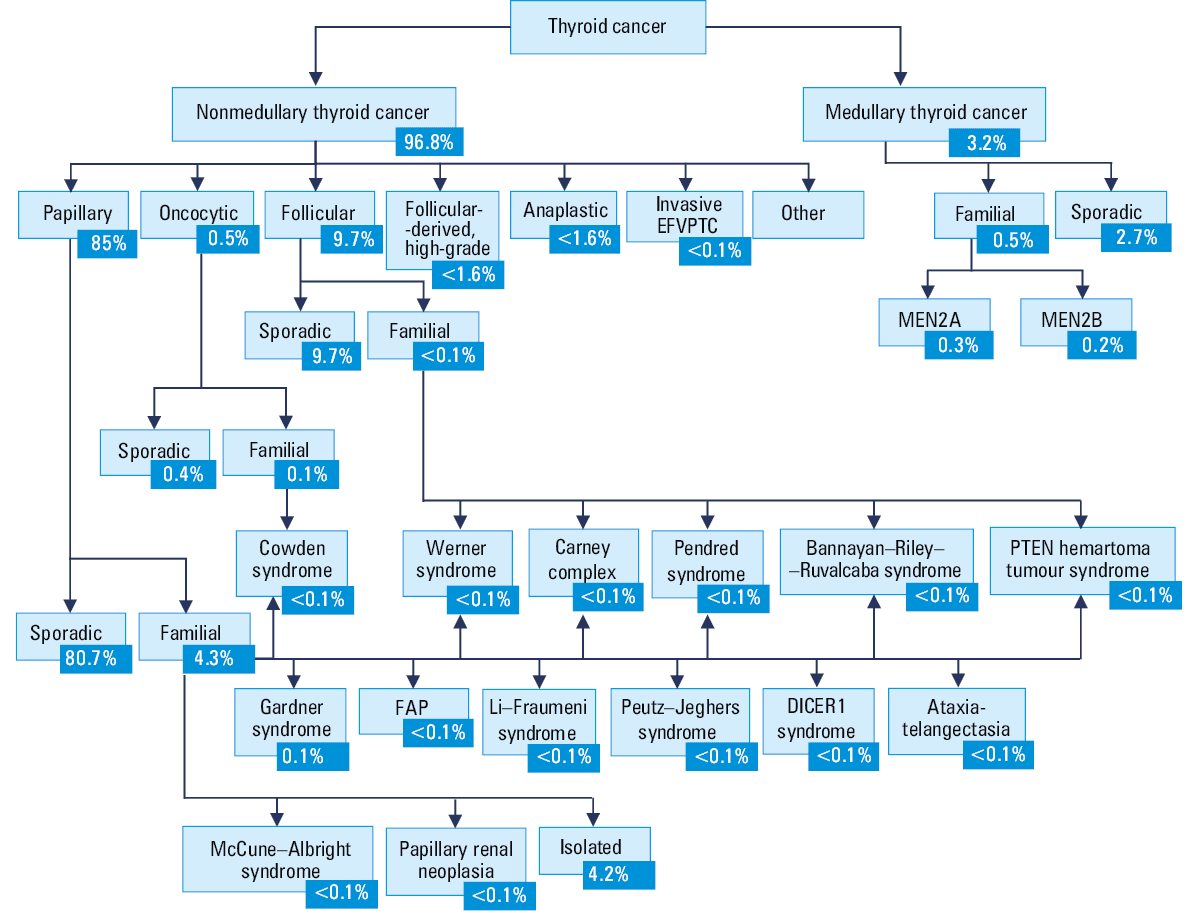
Abbreviations: EFVPTC, encapsulated follicular variant of papillary thyroid carcinoma; FAP, familial adenomatous polyposis; MEN, multiple endocrine neoplasia
In the last 30 years, availability of genome sequencing brought about progress in elucidating the molecular mechanisms underlying TC.15 However, most studies involved somatic mutation analysis from freshly frozen, paraffin‑embedded samples from thyroidectomy.16 Driver mutations that promote cancer development were identified in over 90% of TCs,17 but germline mutations showing genetic susceptibility to DTC are less studied. For the latter, genetic studies are vital. Despite completion of the Human Genome Project,18 information on the full spectrum of human genetic variation remains incomplete.19 Open, population‑scale databases of human genetic variation are important for clinical genetics, biomedical research, prioritizing and tailoring genetic screening programs, or improving guidelines for genetic counselling.20
High‑quality sequencing data enabled building of a unique repository of genetic variation in the Polish population, released publicly as the Thousand Polish Genomes database.20 This database includes small and structural variants, runs of homozygosity, mitochondrial haplogroups, and novel variants identified in the genomes of 1076 Poles.
Genetic background predisposing to DTC may differ among populations. To our best knowledge, there is no report including the Slavic population, which accounts for over 4.5% of the world’s inhabitants. The Polish population, which is homogenous and sedentary in its nature but influenced by many migrations of the past, is not unique and seems highly similar to many other European populations. Therefore, it can serve as a genetic reference for the Slavic populations as long as there are no broader studies from other countries.20
The risk of TC inheritance may be 8- to 12‑fold higher for the first‑degree relatives, as compared with the general population.21 It makes TC one of the most heritable cancers displaying Mendelian inheritance.15 A comparison of the disease aggressiveness in FNMTC patients and sporadic cases yields ambiguous results.15 FNMTC may present a more aggressive disease course at a younger age, with larger tumors and greater lymph node involvement.22 FNMTC may also express clinical anticipation with presentation at a younger age, with more severe symptoms in the second generation (genetic anticipation).23 Early diagnosis based on screening enables identification of TCs of smaller size with fewer lymph node metastases and thus requiring less extensive treatments, potentially improving the treatment outcome.24 Penetrant mutations in FNMTC susceptibility genes could be vital for identifying at‑risk individuals, thereby enabling early diagnosis and selection of appropriate treatment.15 Genetic heterogeneity in the risk assigned to particular susceptibility genes or / and significant differences in the risk allele frequencies require genetic screening matched to the relevant population. Therefore, the aim of the study was to assess the prevalence of germline mutations predisposing to FNMTC in the cohort of Polish individuals, and to compare prevalence of these mutations with the non‑Finnish European (NFE) population.
Patients and methods
Gene search strategy
The first step was to define the list of genes, for which germline mutations have already been linked to increased risk of developing DTC. Our search strategy included Medical Subject Headings terms and keywords: “familial” OR “hereditary” AND “nonmedullary thyroid cancer”. Reference lists of all the selected articles, previous meta‑analyses, and reviews were hand‑searched for any additional articles. We included studies, regardless of their sample size, that investigated the association between germline mutations and DTC occurrence. We carried out a systematic review following the guidelines formulated in the Cochrane Handbook for Systematic Reviews of Interventions and the Preferred Reporting Items for Systematic Reviews and Meta‑Analyses (PRISMA) guidelines13. We searched the following databases: PubMed, MEDLINE, Academic Search Complete, CINAHL Complete, CINAHL, Scopus, Cochrane, Health Source: Nursing / Academic Edition, Web of Knowledge, MasterFILE Premier, Health Source‑Consumer Edition, Agricola, Dentistry, and Oral Science Source from January 2006 up to January 2023 to find all relevant, full‑text journal articles written in English.
Data extraction and methodology assessment
Two authors (MBorowczyk and MBraszka) independently selected the publications that met the inclusion criteria specified above and extracted data for the outcomes using a standardized data extraction form. The risk of bias in the included studies was independently assessed based on the Cochrane risk of bias tool. All included studies were assessed using the Newcastle–Ottawa Scale.13 Only the studies rated with at least 7 stars were included in further analysis. Abstracts / papers focusing on somatic mutations were excluded. The final list of NMTC susceptibility genes was identified and divided into genes related to syndromic and nonsyndromic FNMTC.
Genetic study
The cohort analyzed in this study consisted of 1076 unrelated individuals of Polish origin. Full cohort description was provided earlier.20 Written informed consent was obtained from all participants. Median (interquartile range [IQR]) age of the participants was 47 (35–60) years, with predominance of men (619 vs 457). The analysis of clinical data showed that the most common chronic diseases reported by the participants were hypertension (13%), cancer (4.6%), diabetes (4%), and hypothyroidism or Hashimoto disease (3%). No health problems (excluding COVID‑19) were reported by 86% of the individuals. The sequence data encompassed over 1018 billion read pairs, yielding an average 35.26 × read depth per genome. In every sample, over 91% of the reference genome was covered with at least 10 reads. The allelic frequencies of small and structural variants identified in this cohort were released openly for academic and clinical research as the Thousand Polish Genomes database.20
The list of genes selected for further analysis (Figure 2 and Table 1) was based on the review of previous studies and included 90 genes involved in the development of DTC. The variants were annotated using the following resources: Ensemble Variant Effect Predictor v. 108, including references to databases of genomic variants from ClinVar v. 20221224, and dbSNP build 154, variant population frequencies from the Thousand Polish Genomes Project, and gnomAD v. 2.0.1 and v. 3.0, as well as pathogenicity scores, such as Polyphen‑2, SIFT, and CADD. All gene coordinates were padded with variants in the 10 kb range at both ends of the genes. The analyzed set contained 165 736 variants in 90 genes. Following the ClinVar classification, we filtered pathogenic and potentially pathogenic variants for further analysis. The frequency of variants in the Polish cohort was compared with the frequency estimated for the NFE population, obtained from the gnomAD database (accessed on January 19, 2023).

Abbreviations: FNMTC, famillial nonmedullary thyroid cancer; WGS, whole genome sequencing
Name | Gene | Mode of inheritance | Thyroid cancer histologic subtype | Phenotypes other than thyroid cancer |
Abbreviations: ATC, anaplastic thyroid cancer; DTC, differentiated thyroid cancer; FTC, follicular thyroid cancer; iEFVPTC, invasive encapsulated follicular variant of papillary thyroid carcinoma; PTC papillary thyroid cancer | ||||
FAP and Gardner syndrome | APC | Autosomal, dominant | PTC | Colorectal carcinoma, ampullary carcinoma, hepatoblastoma, medulloblastoma |
Cowden syndrome | PTEN, SDHB‑D, PIK3CA, AKT1, KLLN, SEC23B | Autosomal, dominant | PTC, iEFVPTC,
FTC | Multiple hamartomas, follicular thyroid carcinoma, benign thyroid nodules, breast cancer, endometrial cancer |
Werner syndrome | WRN | Autosomal, recessive | PTC, FTC,
ATC | Premature aging, scleroderma‑like skin changes, cataracts, subcutaneous calcifications, muscular atrophy, diabetes |
Carney complex | PRKAR1 | Autosomal, dominant | PTC, FTC | Spotty skin pigmentation, cardiac myxomas, endocrine tumors |
DICER1 syndrome | DICER1 | Autosomal, dominant | PTC, DTC | Endocrine tumors (thyroid, parathyroid, pituitary, pineal gland, endocrine pancreatic, medullary, adrenocortical, ovarian, and testicular tumors, paragangliomas) |
Pendred syndrome | SLC26A4, FOXI1, KCNJ10 | Autosomal, recessive | PTC, FTC, ATC | Sensorineural deafness / hearing impairment, goiter, abnormal organification of iodide with or without hypothyroidism |
Ataxia‑telangiectasia | ATM | Autosomal, recessive | PTC | Cerebellar degeneration, telangiectasia, immunodeficiency, recurrent sinopulmonary infections, radiation sensitivity, premature aging, lymphoid cancer, poor growth, gonadal atrophy, insulin‑resistant diabetes |
Bannayan–Riley–Ruvalcaba syndrome | PTEN | Autosomal, dominant | PTC, FTC | Macrocephaly, hamartomatous tissue overgrowth, lipomas, pigmented macules on the penis, developmental delay, large birth weight, joint hyperextensibility, endometrial cancer, renal cell carcinoma, Lhermitte–Duclos disease |
Peutz–Jeghers syndrome | STK11 | Autosomal, dominant | PTC, DTC | Gastrointestinal polyposis, mucocutaneous pigmented macules, breast cancer, uterine cancer, cervical cancer, lung cancer, ovarian cancer, testicular cancers |
PTEN hamartoma tumor syndrome | PTEN | Autosomal, dominant | FTC, PTC,
iEFVPTC | Breast cancer, endometrial cancer, gastrointestinal hamartomas, Lhermitte–Duclos disease, macrocephaly, macular pigmentation of the glans penis, multiple mucocutaneous lesions, autism spectrum disorder, colon cancer, esophageal glycogenic acanthosis, lipomas, mental retardation, renal cell carcinoma, testicular lipomatosis, thyroid adenoma, multinodular goiter |
Li–Fraumeni syndrome | TP53 | Autosomal, dominant | PTC, iEFVPTC | Adrenocortical carcinomas, breast cancer, central nervous system tumors, osteosarcomas, soft‑tissue sarcomas, leukemia, lymphoma, gastrointestinal cancers, cancers of head and neck, kidney, larynx, lung, skin, ovary, pancreas, prostate, and testis |
Statistical analysis
Differences in alternative allele frequencies between the Polish cohort and the gnomAD NFE cohort were expressed in terms of odds ratio (OR) defined as
where AFPL is alternative allele frequency in the Polish cohort and AFNFE is alternative allele frequency in the gnomAD NFE cohort. Statistical significance of ORs for each variant was estimated with the Fisher exact test and corrected for false discovery rate, where only the variants with q value below 0.05 were considered significant. All statistical analyses and data visualization were performed with R software (R Foundation for Statistical Computing, Vienna, Austria).
Rechecking data in genetic resources
Variants with significant differences in allele frequency in the Polish population (Thousand Polish Genomes database) and the NFE population (gnomAD database) were clinically annotated using resources such as ClinGen, NCCN guidelines, OMIM, Genetics Home Reference, GeneCards, ClinVar, and Gene‑NCBI to describe their role and possible impact on DTC occurrence.
The study was conducted in accordance with the Declaration of Helsinki, the highest data security standards 140 of ISO 27001, and the General Data Protection Regulation act, and approved by the Ethics Committee of the Central Clinical Hospital of the Ministry of the Interior and Administration in Warsaw (41/2020 and 125/2020).
Results
Among the 90 genes considered, 19 were related to syndromic FNMTC (Table 1) and 71 were related to nonsyndromic FNMTC. Following the ClinVar classification, out of the 165 736 variants located in those genes, we selected pathogenic and potentially pathogenic ones for further analysis. The allele frequencies of variants in the Polish cohort were compared with the frequencies estimated for the NFE population obtained from the gnomAD database. Among them, 23 had significantly different frequencies in the Polish population than in the NFE cohort, and they were the APC, ARSB, ATM, BRCA1, CHEK2, DICER1, GPD1L, INSR, KCNJ10, MYH9, PALB2, PLCB1, PLEKHG5, PTEN, RET, SEC23B, SERPINA1, SLC26A4, SMAD3, STK11, TERT, TOE1, and WRN genes. Most of these genetic variants are more frequent in the Polish population, except for DICER1 and KCNJ10.
Among the genes related to syndromic FNMTC (Table 1), those significantly characteristic for the Polish population and with the highest OR were APC (rs201375478; OR, 22.99), ATM (rs3092859; OR, 13.55), DICER1 (rs117358479; OR, 4.93), KCNJ10 (rs145947380; OR, 11.76), SEC23B (rs138198461; OR, 4.77), SLC26A4 (rs17154362; OR, 15.55), STK11 (rs587782259; OR, 24.31), and WRN (rs4987238; OR, 5.65) (Table 2, Supplementary material, Table S1). Among the genes related to nonsyndromic FNMTC (Table 1), the ones significantly characteristic for the Polish population and with the highest OR were ARSB (rs200040980; OR, 8.62), BRCA1 (rs80357087; OR, 17.02), CHEK2 (rs121908698; OR, 47.42), MYH9 (rs762239398; OR, 126), PALB2 (rs377085677; OR, 21.08), PTEN (rs180953647; OR, 14.47), RET (rs377767388; OR, 50.59), and TERT (rs377216965; OR, 42.15). The genetic variants with an impact as modifiers according to the ClinVar were ARSB (rs72764913), ATM (rs879796523), DICER1 (rs1555368535), KCNJ10 (rs116418256, rs192835895, rs56656397), PALB2 (rs138200248), PCLB1 (rs532302075), PTEN (rs180953647), SERPINA1 (rs11558258), SLC26A4 (rs17154362), SMAD3 (rs958007552), STK11 (rs587782259), and TOE1 (rs3219466).
Gene | Type of thyroid cancer | Hereditary syndromes | Other cancers | Reference |
a Genetic variants without increased frequency in the Polish population
Abbreviations: N/A, not applicable; others, see Table 1
References S1 to S20 are listed in Supplementary material. | ||||
APC | PTC | FAP and Gardner syndrome | Colorectal cancer, ampullary carcinoma, hepatoblastoma, medulloblastoma | Kamani et al15 (2022) |
ARSB | DTC | N/A | N/A | Figlioli et al27 (2014) |
ATM | PTC | Ataxia‑telangiectasia | Cerebellar degeneration, telangiectasia, immunodeficiency, recurrent sinopulmonary infections, radiation sensitivity, premature aging, lymphoid cancer, poor growth, gonadal atrophy, insulin resistant diabetes | Kamani et al15 (2022) |
PTC, DTC | N/A | N/A | Dombernowsky et alS1 (2008) | |
BRCA1 | PTC | N/A | N/A | Wójcicka et alS2 (2014) |
CHEK2 | Nonsyndromic DTC | N/A | Breast cancer, prostate cancer | Wang et alS3 (2019) |
PTC | N/A | N/A | ||
DICER1a | PTC, DTC | DICER1 syndrome | Endocrine tumors (parathyroid, pituitary, pineal gland, endocrine pancreatic, medullary, adrenocortical, ovarian, and testicular tumors, paragangliomas) | Rutter et al32 (2016) |
N/A | Nephroblastoma, NMTC pleuropulmonary blastoma, cystic nephroma, multinodular goiter, thyroid adenoma, sex cord tumor | Zhou et alS4 (2021) | ||
GPD1L | DTC | N/A | N/A | Figlioli et alS5 (2014) |
INSR | PTC | N/A | N/A | Son et al33 (2017) |
FTC | N/A | N/A | Lai et alS6 (2017) | |
KCNJ10 a | ATC | Pendred | Sensorineural deafness / hearing impairment, goiter, and an abnormal organification of iodide with or without hypothyroidism | Liu et alS7 (2016) |
MYH9 | PTC, FTC | N/A | N/A | Wang et al35 (2017) |
PALB2 | PTC | N/A | N/A | Kamihara et alS8 (2022) |
PLCB1 | PTC | N/A | N/A | Bakhsh et alS9 (2018) |
PLEKHG5 | PTC | N/A | N/A | Sarquis et al34 (2020) |
PTEN | PTC, iEFVPTC, FTC | Cowden syndrome | Multiple hamartomas, follicular thyroid carcinoma, benign thyroid nodules, breast cancer, endometrial cancer | Bevan et alS10 (2001) |
PTC, FTC | Bannayan–Riley–Ruvalcaba syndrome | Macrocephaly, hamartomatous tissue overgrowth, lipomas, pigmented macules on the penis, developmental delay, large birth weight, joint hyperextensibility, endometrial cancer, renal cell carcinoma, Lhermitte–Duclos disease | ||
FTC, PTC, iEFVPTC | PTEN hamartoma tumor syndrome | Breast cancer, endometrial cancer, FTC, gastrointestinal hamartomas, Lhermitte–Duclos disease, macrocephaly, macular pigmentation of the glans penis, multiple mucocutaneous lesions, autism spectrum disorder, colon cancer, esophageal glycogenic acanthosis, lipomas, mental retardation, renal cell carcinoma, testicular lipomatosis, PTC, fvPTC, thyroid adenoma, MNG | ||
RET | PTC | FAP | Colorectal carcinoma, ampullary carcinoma, hepatoblastoma, medulloblastoma | Cetta40 (2015) |
SEC23B | PTC, iEFVPTC,
FTC | Cowden syndrome | Multiple hamartomas, follicular thyroid carcinoma, benign thyroid nodules, breast cancer, endometrial cancer | Yehia et alS11 (2015) |
SERPINA1 | PTC | N/A | N/A | Vierlinger et alS12 (2011) |
SLC26A4 | FTC, DTC | Pendred syndrome | Sensorineural deafness / hearing impairment, goiter, and abnormal organification of iodide with or without hypothyroidism | Makhlouf et alS13 (2016) |
SMAD3 | DTC | N/A | N/A | Gudmundsson et alS14 (2017) |
PTC | N/A | N/A | Zhang et alS15 (2014) | |
STK11 | PTC, DTC | Peutz–Jeghers syndrome | Gastrointestinal polyposis, mucocutaneous pigmented macules, breast cancer, uterine cancer, cervical cancer, lung cancer, ovarian cancer, testicular cancers | Buryk et alS16 (2015) |
TERT | DTC | N/A | N/A | Gudmundsson et alS14 (2017) |
PTC | N/A | N/A | Kim et alS17 (2022) | |
ATC | N/A | N/A | Abe et alS18 (2021) | |
TOE1 | PTC | FAP | Colorectal carcinoma, ampullary carcinoma, hepatoblastoma, medulloblastoma | Landrum et alS19 (2016) |
WRN | PTC, FTC,
ATC | Werner syndrome | Premature aging, scleroderma‑like skin changes, cataracts, subcutaneous calcifications, muscular atrophy, diabetes | Lauper et alS20 (2013) |
Figure 3 shows allele frequencies of variants in 25 genes with significantly different allele frequency between the Polish and gnomAD NFE cohorts. Table 2 describes genes with significant differences in allele frequencies in these 2 cohorts.
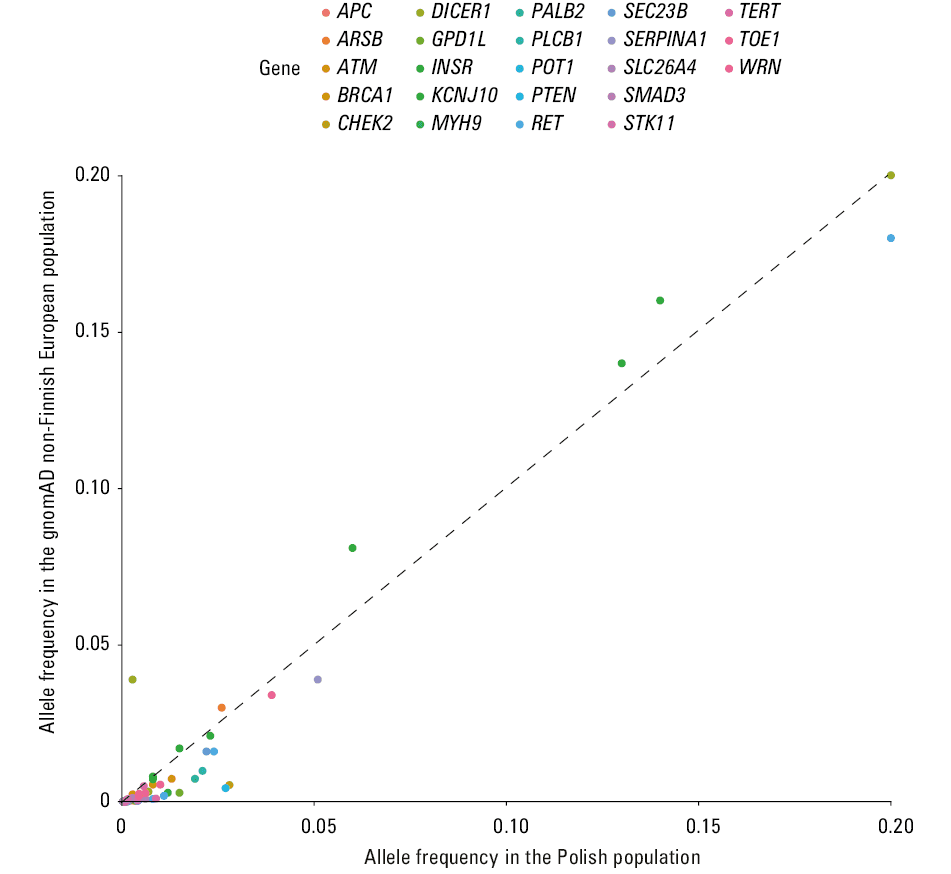
Abbreviations: see Table 1
Discussion
Between 5% and 15% of NMTC cases occur due to germline mutations.22 Despite mounting evidence of at least partial TC heritability, to date, only a handful of genetic variants have been convincingly associated with a higher risk of this cancer.15 As studies have shown, high heritability of TC is likely due to the occurrence of many rare but high‑penetrance genetic variants in some cases, or common, low‑penetrance variants in the others.25
Of the genes analyzed in our study, 19 were related to syndromic FNMTC and 71 to nonsyndromic FNMTC. Within this group, significant variants included 23 genes, and indeed many of them have been already described as important in the initiation or progression and development of TC. However, not many of them have ever been described in the context of the population frequency. Most of those genetic variants are more frequent in the Polish population (Thousand Polish Genomes database) than in the NFE population (gnomAD), except for DICER1 and KCNJ10. Below, we briefly discuss each of the genes, and consider their contribution to TC and known prevalence in other populations.
The APC gene variants are well known for their contribution to familial adenomatous polyposis (FAP) and Gardner syndrome; however, papillary thyroid carcinoma (PTC) associated with FAP is very rare.26 The most significant variants occurring more frequently in the Polish population (Supplementary material, Table S1) are missense variants of conflicting interpretation of pathogenicity. All of them have been described before and are known to be cancer‑related.
The expression level of ARSB in the TC tissue is low, but has been reported previously in the Italian population.27 In our cohort, 3 variants reached statistical significance; however, their prevalence is not very different from the other analyzed populations listed in the gnomAD database. This gene has not been described in detail in the context of TC, and may be an interesting novel candidate for further studies.
The ATM gene is crucial when it comes to a response to ionizing radiation–induced DNA damage, and therefore its possible involvement in 2.4% of TCs has been suspected for a long time.28 Mutations of the ATM gene are responsible for ataxia telangiectasia. In our cohort, several variants of the ATM gene have been discovered as significantly more frequent, many of them with a clearly pathogenic status (Supplementary material, Table S1).
BRCA1, similarly to the CHEK2 and PALB2 genes, is engaged in the DNA repair mechanisms, and therefore is crucial for cancer protection. In our cohort, only 3 variants of this gene have been significantly more frequent than in the gnomAD cohort, 2 of them are of pathogenic status, and 1 is a missense variant of conflicting interpretation status. It has been suggested that CHEK2 mutations generally predispose to TC, together with familial aggregations of breast cancer and TC, and even to double primary breast and thyroid cancers, with several cases already described in the Polish population.29 The gene plays a role in maintaining genomic stability, and it also acts as a tumor suppressor.30
The DICER1 gene mutations are crucial for DICER1 syndrome.31 Co‑occurrence of DTC with Sertoli–Leydig cell tumor is highly suggestive of DICER1 syndrome.31 Most patients with DICER1 syndrome diagnosed with DTC (PTC and follicular TC [FTC]) had prior exposure to radiation and chemotherapy for the treatment of associated malignancy.31The risk of DTC was 16- to 24‑times greater over a DICER1 patient’s lifetime.32 In our study, the frequency of DICER1 variants was similar to that of the comparator, except for 1 variant with slightly higher prevalence in the Polish population, but not clearly pathogenic (Supplementary material, Table S1).
GPD1L mutations in patients with DTC were described in the Italian, Polish, and Spanish populations,27 whereas INSR mutations were only reported in the Korean population.33 In our cohort, 1 variant of the GPD1L gene and all analyzed INSR variants occurred statistically more frequently than in the gnomAD control group, with a status of conflicting interpretation of pathogenicity (Supplementary material, Table S1).
KCNJ10 mutations were described in patients with Pendred syndrome.15 Little is known about the frequency of its variants in other populations; however, in our cohort at least 8 variants seem important (Supplementary material, Table S1). Interestingly, 5 of them are in the 3' untranslated region (UTR) of the gene.
PLEKHG5 germline variants in familial nonsyndromic PTC (classic and follicular variant) were identified in the Brazilian population.34 In our study, 1 missense variant was found to be significantly more frequent in the Polish population.
MYH9 encodes myosin‑9 and potentially affects the risk of PTC by interacting with a long noncoding RNA (encoded by the PTCS2 gene) and the FOXE1 gene.35 Only 1 missense variant has been found to be significantly more frequent in the Polish cohort, even though it has a status of conflicting interpretation of pathogenicity. It may also be an interesting candidate gene for further studies on DTC. This gene is a novel cancer stem cell marker claimed as a prognostic indicator in esophageal cancer that promotes oncogenesis through the PI3K/AKT/mTOR axis.36
The InDel intronic variant within PLCB1 was the first mutation identified in familial multiple papilloid adenomata‑type DTC patients and in a subset of patients with sporadic DTC.37 In patients who were carriers of this mutation, multinodular goiter progressed to PTC (follicular variant) through overexpression of phospholipase C-β1.38 In the Polish population, 3 variants reached statistical significance with higher prevalence but conflicting interpretation of pathogenicity.
Approximately 6% to 38% of PTEN hamartoma tumor syndrome patients develop TC at a median age of 31–37 years.15 A risk of DTC is 26 to 39 times higher than for individuals without a PTEN mutation.39 In our study, 3 variants located in the noncoding regions have been found more frequently in the Polish population; however, little is known about them in the context of DTC.
Although the RET gene mutations are crucial for medullary TC development, a germline RET oncogene mutation may be also linked to NMTC, especially as a part of FAP syndrome.40 In our study, several genetic variants of this gene have been found as being significantly more frequent in Poles. Most of them are missense variants or are located in the 3'UTRs or 5'UTRs.
A SEC23B mutation was identified in patients with FTC as a component of the Cowden syndrome in a whole genome sequencing (WGS) study.41 In the carriers of the SEC23B pathogenic variant, Yehia et al41 found a significantly increased age‑adjusted standardized incidence rate of DTC, as compared with the general population. Again, in our population, the discovered variants are more frequent, with 1 of them possessing a pathogenic status (Supplementary material, Table S1).
SERPINA1 (α1‑antitrypsin) is highly expressed in papillary TC.34 A role of this gene in thyroid tumorigenesis is not fully understood. Its mutations may reflect mitogenic activity, stimulating malignant cell proliferation.34 One variant of this gene located in the 5'UTR region has been found as statistically more prevalent in the Polish population.
The SLC24A6 gene, which encodes a mitochondrial sodium and calcium ion exchanger, was found to considerably affect the risk of developing TC.33 However, its relationship with DTC pathogenesis has not been established yet, even though an interesting finding was a stronger association of its genetic variant with FTC rather than with PTC in a Korean study.44 This striking finding suggested that at least some genetic markers of susceptibility to DTC are different between TC types. In our study, many variants seem to be more frequent in the analyzed population, most of which are missense variants with conflicting interpretation of pathogenicity, whereas 1 discovered variant is likely pathogenic as a frameshift mutation.
The SMAD family member 3 gene (SMAD3) shows higher expression in the thyroid than in most other tissues. This supports its potential role in predisposition to TC.11 SMAD3 is an important transcriptional mediator of transforming growth factor-β (TGF-β) signaling associated with PTC.15 Wang et al43 investigated downstream mechanisms in which alterations of SMAD3 contribute to TC susceptibility. Only 2 variants of conflicting interpretation of pathogenicity have been found in our study as being slightly more prevalent in the Polish cohort.
A mutation in the serine / threonine kinase 11 (STK11) gene is a causative agent of Peutz–Jeghers syndrome (PJS).15 An increased cancer risk has been related to the P53 pathway.44 PJS has been associated with multiple cases of PTC and FTC.15 Only 1 intronic variant has been significantly more frequent in the Polish cohort, and it is located in the intronic region of the gene, thus, it may possess a regulatory function.
The TOE1 gene product inhibits cell growth rate and cell cycle, as it induces the CDKN1A gene expression and TGF-β expression. Moreover, it also mediates EGR1-induced growth inhibition.45 TOE1 mutations were associated with PTC occurring in the patients with FAP. In our study, 1 intronic variant seems to be slightly more prevalent in the Polish population, as compared with the gnomAD cohort, but currently it has a conflicting interpretation of pathogenicity.
The TERT gene encodes a protein that is an essential component of the telomere length maintenance complex and is weakly expressed in normal thyroid tissue. However, it is known to be reactivated in many human cancers, including TC, through a transcriptional regulation.42 It is correlated with a more severe form of the cancer disease, and mutations in the TERT promoter in TC were suggested as having a prognostic potential if coexisting with the BRAFV600E mutation.43 In our study, 2 variants of the TERT gene have been found as occurring more frequently in the Polish population, both with conflicting interpretation of pathogenicity.
Mutations in the WRN gene are associated with Werner syndrome,47 and they may increase the cancer risk irrespective of their location. Several missense variants have been found to occur more frequently in our cohort than in the control gnomAD cohort; however, all of them with conflicting interpretation of pathogenicity.
The population studies are crucial, since in every cohort there might be several founder variants typical of a given population but absent or very rare in other groups. Some genetic variants may be more prevalent in many populations due to historical influences. This may be valuable for clinical geneticists and other health care professionals, as they may provide us with additional guidelines at the time of diagnosis. Due to financial constraints, genetic testing is still not very popular in many countries, and an analysis of selected variants or genes, instead of huge panels or WGS, is simply cheaper and faster, and therefore, more available.
Despite our best efforts, there are several limitations to the study. First, we decided to focus only on the protein‑coding genes, since the knowledge about other elements of the genome, especially those affecting the TC risk, is still in its infancy. Only 2 studies indicated an association of PTC with mutations within miRNA genes in the Polish population.47,48 Secondly, a reliable comparison of allele frequency between populations suffers from a low number of available data. The gnomAD cohort contains data for participants from many different populations, but usually of a small sample size that does not allow for an accurate allele frequency estimation. Especially when it comes to the WGS data, there are still only a few large populational databases. Despite interesting findings, it is important to stress that only carefully selected genes have been analyzed in our research. Even if the entire gene sequence was taken into consideration, there might still be other elements of the genome with equal importance or even contributing more to the regulation of gene expression. Also, epigenetic factors are known to be crucial at many cancer stages, and further studies are necessary to fully understand the TC intricacies.
Conclusions
Despite advances in elucidating the molecular mechanisms underlying DTC development related to genome sequencing, germline mutations responsible for genetic susceptibility to DTC are still poorly recognized. The aim of this study was to assess the prevalence of genetic germline mutations predisposing to DTC in the cohort of Polish individuals, using the whole genome analysis to form the foundations of further tailored prophylaxis. Undoubtedly, additional research is needed to provide a more extensive background on the penetrance, molecular function, and functional consequences of the genetic variants presented here. Such studies can further clarify the etiology of TC and probably support identification of the disease risk in family members of NMTC patients, not solely in the population, but serving as guidelines for further populational studies. Finally, many studies describing the genetic predisposition to NMTC are of a case‑control design. Genome‑wide association studies, preferably based on the next generation sequencing and family‑based exome sequencing are needed. Genomic studies should be followed by transcriptome (eg, single‑cell RNA sequencing) and / or molecular (droplet digital polymerase chain reaction or Sanger sequencing) analyses and linkage studies to identify and confirm new susceptibility loci associated with NMTC.49
- Wells SA. Progress in endocrine neoplasia. Clin Cancer Res. 2016; 22: 4981‑4988. | Crossref
- Deng Y, Li H, Wang M, et al. Global burden of thyroid cancer from 1990 to 2017. JAMA Netw Open. 2020; 3: e208759. | Crossref
- Davies L, Morris LGT, Haymart M, et al. American Association of Clinical Endocrinologists and American College of Endocrinology disease state clinical review: the increasing incidence of thyroid cancer. Endocrine Practice. 2015; 21: 686‑696. | Crossref
- Megwalu UC, Moon PK. Thyroid cancer incidence and mortality trends in the United States: 2000‑2018. Thyroid. 2022; 32: 560‑570. | Crossref
- Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021; 71: 209‑249. | Crossref
SUPPLEMENTARY MATERIAL
ARTICLE INFORMATION