Extreme soft tissue involvement preceding multiple myeloma diagnosis: is Occam’s razor always the answer?
 ,
,

 CC BY 4.0
CC BY 4.0
Extreme soft tissue involvement preceding multiple myeloma diagnosis: is Occam’s razor always the answer?
A 77‑year‑old man was admitted to our department of nephrology due to acute kidney injury. His past medical history included Gleason grade 4+3 prostate cancer treated with robotic prostatectomy. At present, he was being diagnosed for a quickly growing tumor (Figure 1A) that first appeared on the right side of his chest wall around 3 months before the hospitalization, and was described by the patient and his wife as being the size of a tangerine. At that time, he was diagnosed in another hospital, where a computed tomography scan of the chest was performed and revealed a massive soft tissue tumor measuring 90 mm × 125 mm × 100 mm with infiltration and destruction of the sternum body, ribs, and pectoral muscles (Figure 1B). A similar mass measuring 65 mm × 80 mm × 70 mm was seen in the upper mediastinal region. Biopsy of the mass was performed, and a preliminary diagnosis was Ewing sarcoma.
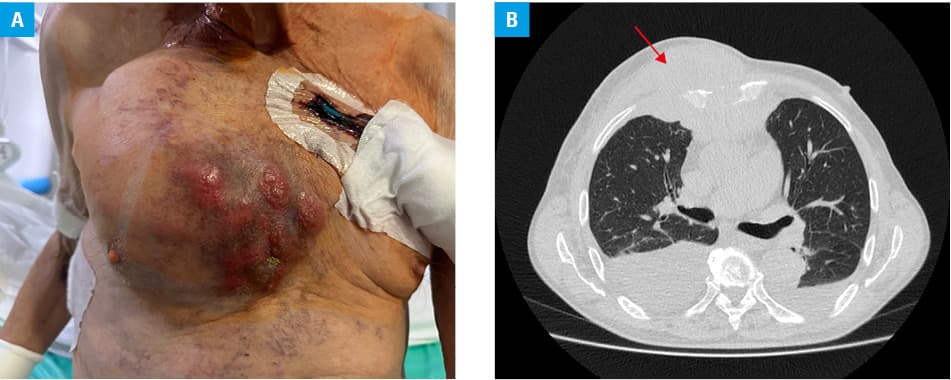
The patient complained of chest wall pain and dyspnea. Physical examination was notable for massive edema. His creatinine level on admission was 598.6 µmol/l (reference range [RR], 64.55–103.45 µmol/l), while a month before the presentation it was 155.62 µmol/l. The other parameters on admission were as follows: uric acid, 820.82 µmol/l (RR, 208.18–428.26 µmol/l), potassium, 5 mmol/l (RR, 3.5–5.1 mmol/l), calcium, 2.23 mmol/l (RR, 2.1–2.55 mmol/l), phosphate, 2.31 mmol/l (RR, 0.74–1.52 mmol/l), and hemoglobin, 10.9 g/dl (RR, 14–18 g/dl). Normal size kidneys without any evidence of obstruction were visualized on ultrasound.
A presumptive diagnosis of tumor lysis syndrome was made. The patient received rasburicase without a clinically significant effect, and was started on hemodialysis. A monoclonal peak was observed on serum protein electrophoresis, and immunofixation together with free light‑chain concentration measurement were performed. The assay revealed extreme λ light‑chain elevation (free λ light‑chains, 7919.02 mg/l; RR, 5.71–26.3 mg/l; free κ light‑chains, 37.48 mg/l; RR, 3.3–19.4 mg/l; κ/λ ratio, 0; RR, 0.26–1.65), therefore, light‑chain cast nephropathy was suspected, and the patient was consulted by hematologists. Bone marrow aspirate and biopsy were obtained together with another tumor biopsy to finally confirm whether the patient had 2 distinct diseases or these were soft tissue plasmocytomas. High‑dose dexamethasone was started immediately pending the arrival of results. The tumor biopsy confirmed the presence of plasma cells with CD138 expression and absence of panCK, synaptophysin, S100, and CD99. Bone marrow core biopsy was characteristic of multiple myeloma (MM) with 30% of clonal plasma cells. Unfortunately, the patient contracted COVID‑19 and died before chemotherapy was started.
Extramedullary involvement in MM represents an aggressive form of the disease characterized by the ability of the clone to grow outside of the bone marrow. Typically, at diagnosis plasmacytomas are found in the skin and soft tissues, and are present in 0.5% to 4.8% of patients.1 There are 3 ways soft‑tissue plasmocytomas can develop, that is, through hematogenous spread, by direct growth from bone tumors, and rarely after invasive procedures.2,3
The presented case not only demonstrates extreme soft tissue involvement in the course of MM, but also perfectly illustrates Occam’s razor or the principle of parsimony in medicine—a unifying diagnosis should be favored over multiple ones in a situation where all of them explain the clinical symptoms equally well.4
- Blade J, Beksac M, Caers J, et al. Extramedullary disease in multiple myeloma: a systematic literature review. Blood Cancer J. 2022; 12: 45. | Crossref
- Rosinol L, Beksac M, Zamagni E, et al. Expert review on soft‑tissue plasmacytomas in multiple myeloma: definition, disease assessment and treatment considerations. Br J Haematol. 2021; 194: 496‑507. | Crossref
- Rosinol L, Fernandez de Larrea C, Blade J. Extramedullary myeloma spread triggered by surgical procedures: an emerging entity? Acta Haematol. 2014; 132: 36‑38. | Crossref
- Kelly J. The diagnostic approach in complex patients: parsimony or plenitude? Am J Med. 2021; 134: 11‑12. | Crossref
ARTICLE INFORMATION