Unveiling the enigma of primary angiosarcoma of the adrenal gland: a rare and aggressive malignancy



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Unveiling the enigma of primary angiosarcoma of the adrenal gland: a rare and aggressive malignancy
Primary angiosarcoma of the adrenal gland is an ultrarare and aggressive malignancy with challenging diagnosis and treatment. Since 1988, only about 50 cases of this neoplasm have been documented.1,2 In addition, adrenal angiosarcoma accounts for less than 1% of all sarcomas.1,2
We present a case of a 69‑year‑old woman with abdominal pain. Ultrasound revealed an 8‑cm lesion in the right adrenal area. A contrast‑enhanced abdominal computed tomography (CT) scan was performed and confirmed a mass of 83 mm × 69 mm × 75 mm in the right adrenal gland. It appeared heterogeneous with fluid‑filled areas, microcalcifications, and enhancement after the contrast administration in solid parts. There was an impression of the right lobe of the liver and the right kidney (Figure 1A and 1B). Chest X‑ray did not reveal any abnormalities.
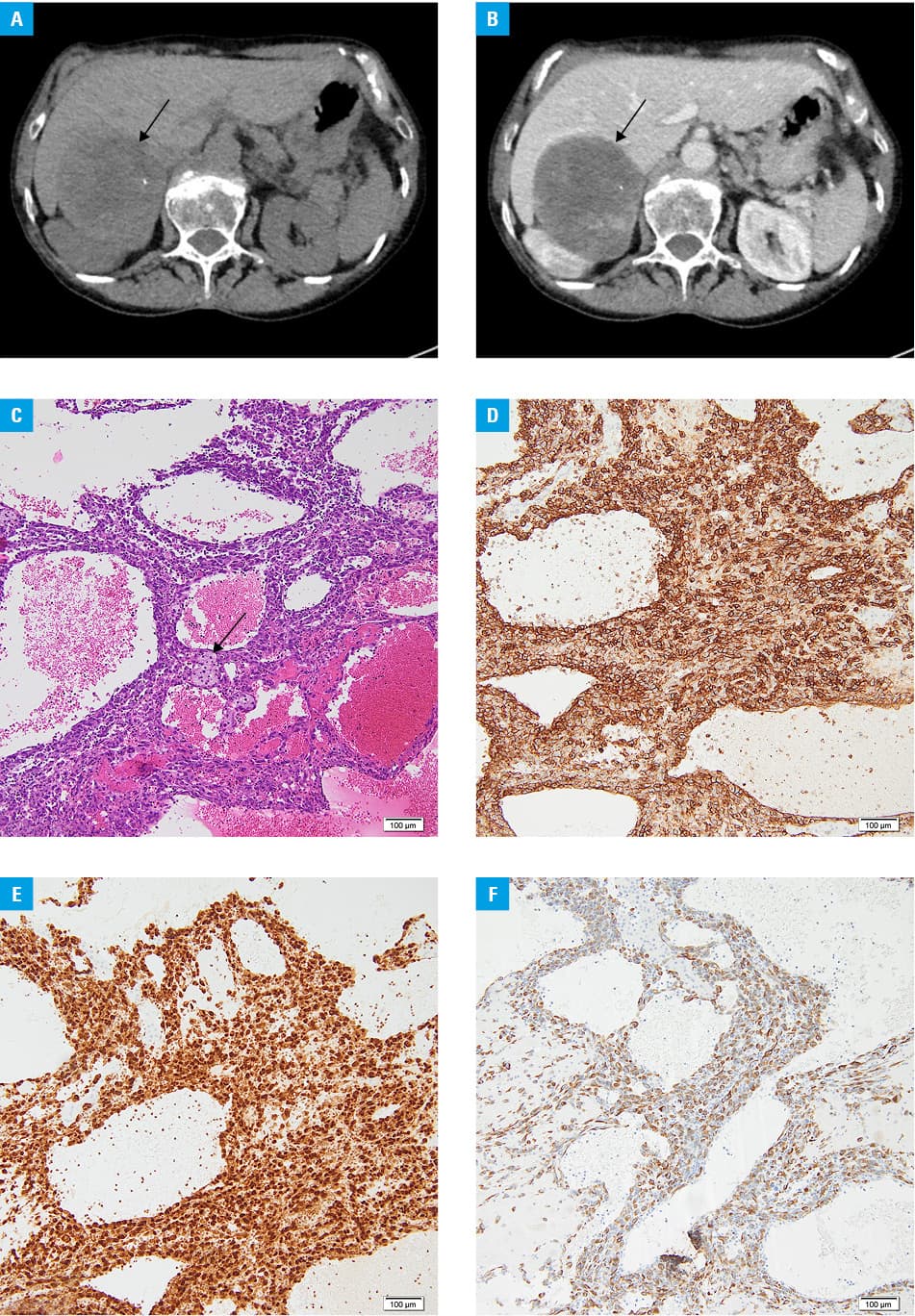
Due to the size and suspicious imaging phenotype of the lesion, the patient was referred for right adrenalectomy, following prior hormonal assessment. Upon admission to an endocrinology department the patient reported no complaints, and physical examination revealed no signs of endocrinopathy. Laboratory tests indicated low adrenal androgen levels, normal levels of renin‑angiotensin‑aldosterone system hormones, normal morning cortisol and adrenocorticotropic hormone levels, as well as normal plasma metanephrine / normetanephrine concentrations. Low‑dose dexamethasone test (1 mg, overnight) revealed incomplete serum cortisol suppression (61 nmol/l; reference range <50 nmol/l). The results imposed steroid coverage for surgery, although there were no indications for α-blockers. Laparoscopic right adrenalectomy was performed without complications, and the patient was discharged home in good general condition.
Histopathologic evaluation revealed angiosarcoma of the adrenal gland. The tumor was mostly hemorrhagic and necrotic (about 85%), composed of epithelioid cells with marked atypia, which formed vascular channels. The mitotic index was 6/10 high‑power fields. The lesion was completely excised with very narrow external margins (0.1 mm) (Figure 1C–1F).
Despite an elevated risk of local recurrence in the adrenal tumor, the patient opted out of adjuvant local radiotherapy. In the following 6 weeks, a CT scan unveiled a local recurrence in the upper segment of the right kidney, measuring 31 mm × 28 mm × 27 mm. This prompted initiation of paclitaxel chemotherapy, though it was discontinued after the initial cycle due to a urinary tract infection. The patient refused further treatment and was provided with hospice care.
Diagnosis of primary adrenal angiosarcoma is challenging. The symptoms reported in the described cases included fever, anemia, weight loss, and chronic pain.1-3 Our patient reported only nonspecific abdominal pain. The CT scans suggested malignant adrenal lesion. The histopathologic evaluation can also be ambiguous. Positive endothelial markers are needed to confirm the diagnosis. There is no standardized treatment protocol for adrenal angiosarcoma. According to current publications, patients with resectable masses should undergo adrenalectomy. If the adjacent organs are infiltrated, their resection is also suggested. Local radiotherapy is often an adjuvant treatment due to the risk of local recurrence. Metastatic angiosarcomas are treated with taxanes, which may suppress angiogenesis of the tumor.4
Effective surgical treatment, adjuvant radiotherapy, and chemotherapy may improve the prognosis of patients with adrenal angiosarcoma; however, the reported 5‑year survival is below 30%.1 Our patient had several poor prognostic factors, including tumor size greater than 5 cm, suspected visceral invasion, tumor necrosis on histopathologic examination, and very short‑time local recurrence.4,5
Certainly, primary angiosarcoma of the adrenal gland requires increased scientific scrutiny to improve understanding and management of the disease.
- Ladenheim A, Tian M, Afify A, et al. Primary angiosarcoma of the adrenal gland: report of 2 cases and review of the literature. Int J Surg Pathol. 2022; 30: 76‑85. | Crossref
- Noman M, Zeer AMM, Zeer ZMM, et al. Epithelioid angiosarcoma of the adrenal gland with metastasis: a case report and literature review. Ann Med Surg (Lond). 2023; 85: 3106‑3112. | Crossref
- Kędzierski L, Hawrot‑Kawecka A, Holecki M, Duława J. Angiosarcoma of adrenal gland. Pol Arch Med Wewn. 2013; 123: 502‑503. | Crossref
- Fury MG, Antonescu CR, Van Zee KJ, et al. A 14‑years retrospective review of angiosarcoma: clinical characteristics, prognosis factors, and treatment outcomes with surgery and chemotherapy. Cancer. 2005; 11: 241‑247. | Crossref
- Buehler D, Rice S, Moody JS, et al. Angiosarcoma outcomes and prognostic factors: a 25‑year single institution experience. Am J Clin Oncol. 2014; 37: 473‑479. | Crossref
ARTICLE INFORMATION