Rapid-onset Cushing syndrome induced by adrenocorticotropic hormone–producing pancreatic neuroendocrine tumor: a therapeutic challenge



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Rapid-onset Cushing syndrome induced by adrenocorticotropic hormone–producing pancreatic neuroendocrine tumor: a therapeutic challenge
Endogenous Cushing syndrome (ECS) results from excessive glucocorticoid secretion. Adrenocorticotropic hormone (ACTH)-secreting pituitary adenomas are the most common presentation, followed by ectopic secretion from lung and thymus neuroendocrine tumors (NETs). Pancreatic NETs rarely cause ECS.1-3 We present a case of rapid‑onset ECS due to malignant pancreatic ACTHoma, resistant to multiple treatment modalities.
A 25‑year‑old man was admitted to a regional hospital in Pleszew, Poland, with new‑onset hypertension and diabetes mellitus. Physical examination revealed central obesity, muscle weakness, and violaceous striae. Laboratory results included hypercortisolemia and hypokalemia. A 45 mm pancreatic tumor was identified on computed tomography (CT) (Figure 1A–1D). Endoscopic ultrasound–guided biopsy confirmed a G2 pancreatic NET (Ki‑67, 15%).
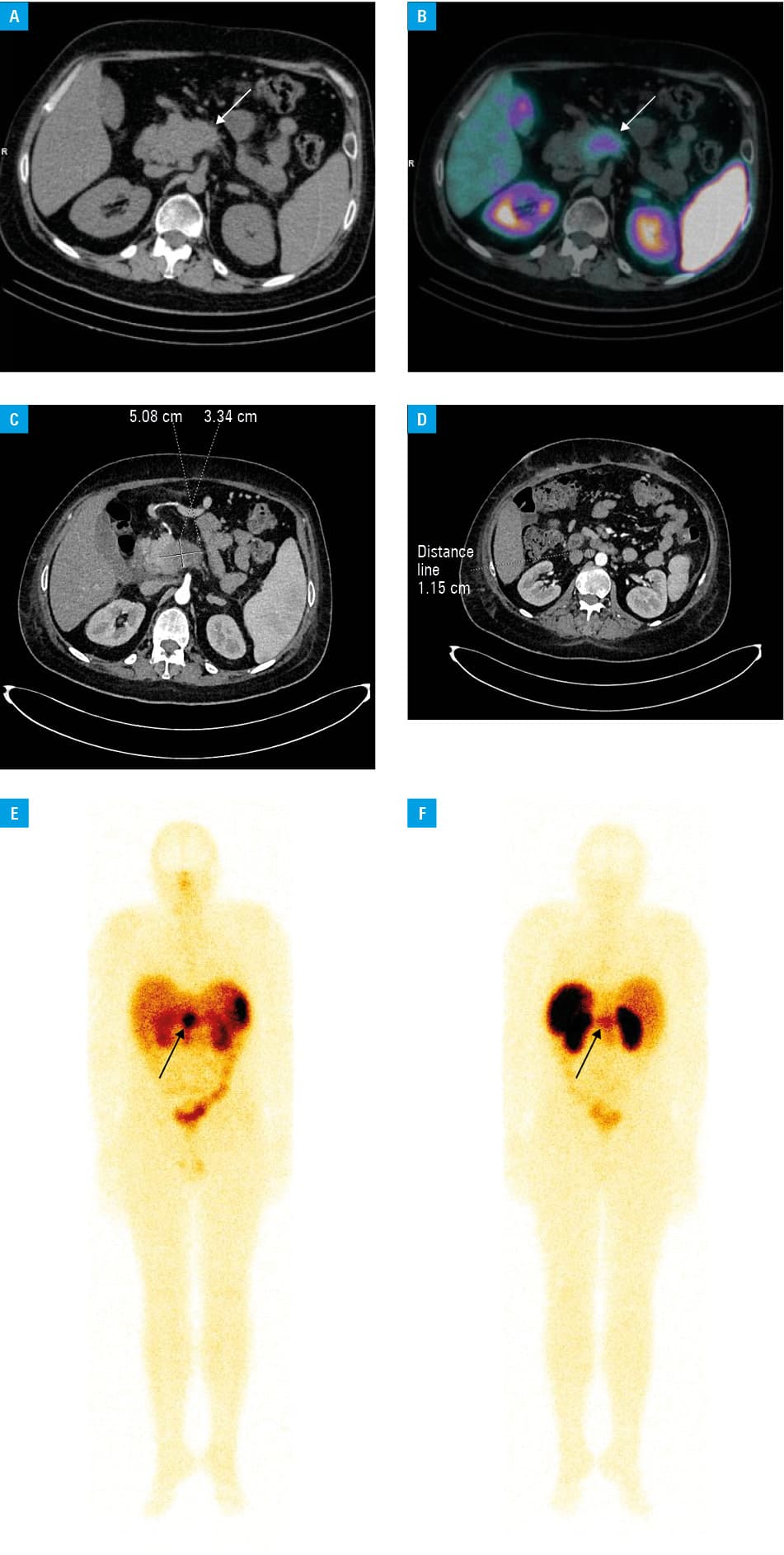
The patient was referred to the Department of Endocrinology in Poznań. Serial monitoring showed disrupted cortisol circadian rhythm, with serum and 24‑hour urine cortisol concentration above 1750 nmol/l (reference range, 166–507 nmol/l) exceeding the assay’s measuring range of 1750 nmol/l. Serum ACTH was markedly elevated at 282.98 pg/ml (reference range, 1.59–13.94 pg/ml), with no decrease after 1 mg and 8 mg dexamethasone suppression tests. Somatostatin receptor overexpression in the tumor tissue was detected on 99mTc‑hydrazinonicotinyl‑Tyr3‑octreotide single photon emission computed tomography / CT. ECS induced by the pancreatic ACTHoma was diagnosed.
The patient was transferred to a tertiary reference center in Warsaw for resection. The surgery was abandoned due to excessive bleeding and tumor necrosis. Wound infection developed, requiring intravenous antibiotics. Perioperative complications resulted from hypercortisolemia, compromising immune response and tissue regeneration, amplified by the tumor’s hypervascularization and tissue infiltration.
Initial treatment with metyrapone failed to reduce cortisol adequately, and osilodrostat was introduced. Despite increasing the dose to 20 mg twice daily, hypercortisolemia persisted. CT showed periaortal lymph node metastases and tumor size progression (51 mm × 34 mm). Compression fractures were found in the lumbar vertebrae on magnetic resonance imaging. The patient’s condition rapidly deteriorated, highlighted by exacerbation of edema, fatigue, worsening physical state (World Health Organization / Eastern Cooperative Oncology Group score of 4), and severe hypokalemia despite continuous intravenous administration of potassium. A tumor board meeting was organized to evaluate treatment options. Ultimately, continuous intravenous etomidate infusion was initiated at the intensive care unit to reduce hypercortisolemia, followed by bilateral adrenalectomy.
Etomidate treatment decreased early‑morning serum cortisol to 615 nmol/l. Due to a risk of complications associated with the presence of ECS and vertebral compression fractures, the patient was disqualified from open adrenalectomy and transferred to the University Hospital in Białystok for videoscopic adrenalectomy. The surgery was successful, with no complications noted. Substitution dose of hydrocortisone was prescribed, achieving an adequate serum cortisol concentration.
The patient continued treatment for nonresectable ACTHoma with lanreotide 120 mg monthly and everolimus 10 mg daily. Due to clinical progression, peptide receptor radionuclide therapy (PRRT) with 177Lu‑DOTA‑0‑Tyr3‑Octreotate was initiated, following a standard regimen of 200 mCi 4 times at 8‑week intervals. Increased radiopharmaceutical uptake in the tumor tissue was visible on postdose scintigraphy (Figure 1E–1F). The patient’s condition stabilized, with no treatment‑related complications.
While an uncommon cause of ECS, pancreatic ACTHomas require consideration. Our report presents a range of therapeutic options and potential challenges in ECS. Steroidogenic enzyme inhibitors are first‑line treatment for nonresectable tumors; however, bilateral adrenalectomy may be warranted in aggressive cases to mitigate hypercortisolemia.4 PRRT is an option in somatostatin receptor–positive NETs.5
- Gadelha M, Gatto F, Wildemberg LE, Fleseriu M. Cushing’s syndrome. Lancet. 2023; 402: 2237‑2252. | Crossref
- Maciejewski A, Gut P, Stajgis P, et al. Combined largecell neuroendocrine carcinoma and atypical carcinoid of the thymus presenting as ectopic Cushing syndrome. Pol Arch Intern Med. 2022; 132: 16312. | Crossref
- Paleń-Tytko JE, Przybylik‑Mazurek EM, Rzepka EJ, et al. Ectopic ACTH syndrome of different origin—diagnostic approach and clinical outcome. Experience of one clinical centre. PLoS One. 2020; 15: e0242679. | Crossref
- Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol. 2021; 9: 847‑875. | Crossref
- Hofland J, Falconi M, Christ E, et al. European Neuroendocrine Tumor Society 2023 guidance paper for functioning pancreatic neuroendocrine tumour syndromes. J Neuroendocrinol. 2023; 35: e13318. | Crossref
ARTICLE INFORMATION