Giant prolactinomas (GPs) are a rare subtype of lactotroph pituitary neuroendocrine tumors that constitute about 3% of prolactinomas. They measure more than 40 mm and occur predominantly in young men.1 The management of GP poses a diagnostic and therapeutic challenge, since literature regarding this condition is scarce and the scale of the problem remains underestimated.
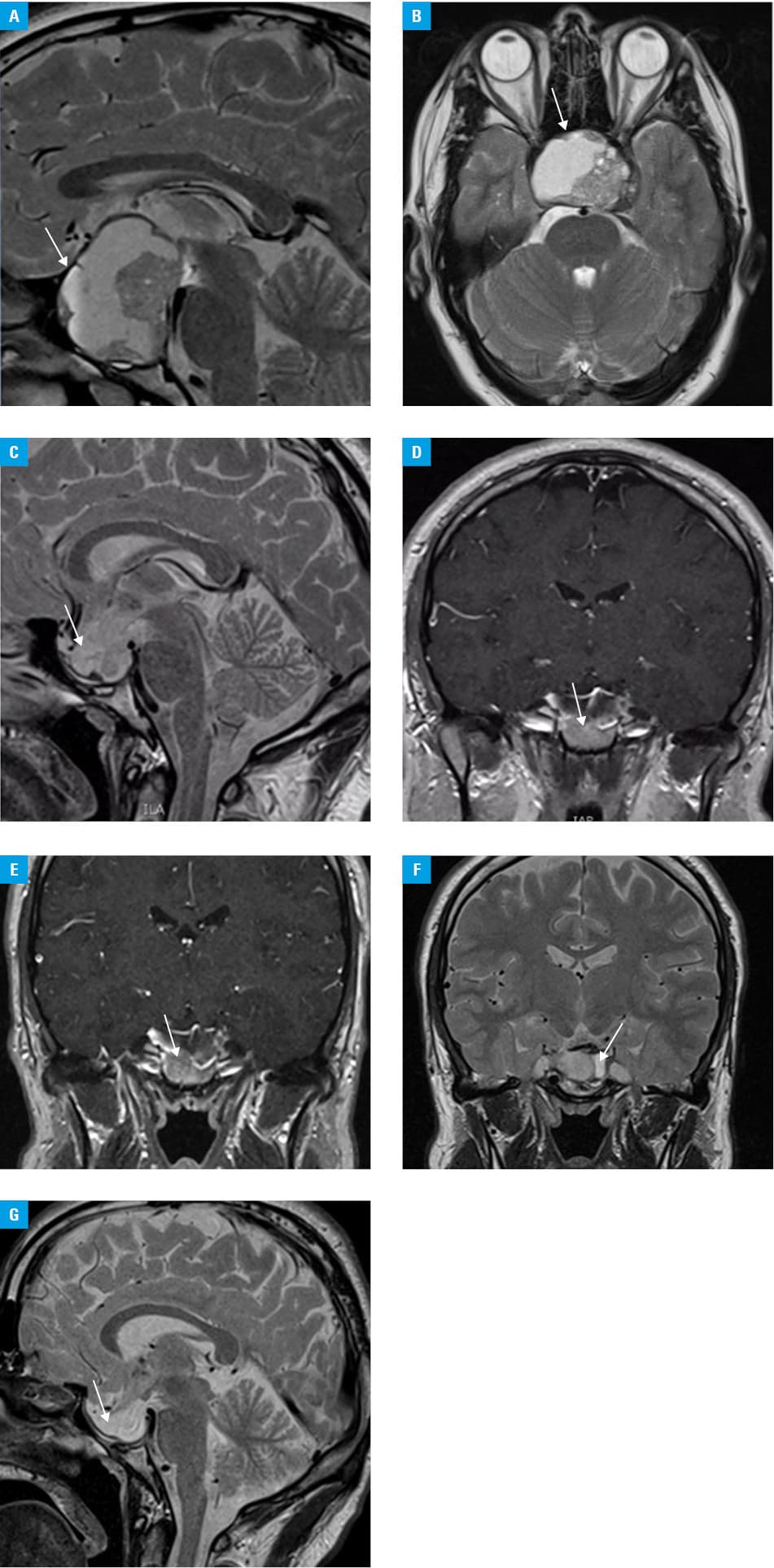
We present a case of a 23‑year‑old man with GP whose incomplete susceptibility to cabergoline should be classified as partially successful. The patient’s 6‑month history included acute headaches and visual deficits. Symptoms of hypogonadism were present since puberty. Magnetic resonance imaging (August 2018) showed a mass (52 mm × 52 mm × 41 mm) with a suprasellar extension, infiltration of the optic chiasm, and a site of previous hemorrhage (Figure 1A and 1B). Considerably elevated serum levels of prolactin (21 525 μlU/ml; reference range, 86–324 μlU/ml) were found with suppressed luteinizing hormone and follicle‑stimulating hormone levels, and lowered level of testosterone. Serum levels of free thyroxine and thyroid‑stimulating hormone indicated central hypothyroidism (Supplementary material, Table S1). Dual‑energy X‑ray absorptiometry showed osteopenia. Ophthalmologic examination detected a bilateral visual field loss in the upper temporal quadrants.
The Pituitary Tumor Board did not find indications for surgery. Pharmacologic treatment with bromocriptine (10 mg/day) was continued, leading to a decrease in prolactin levels (by 63%) and slight headache alleviation. Levothyroxine (75 μg daily), testosterone injections (once daily for 2 weeks), and cholecalciferol (4000 IU daily) were introduced.
Follow‑up examinations (November 2018, May 2019) disclosed further prolactin level decrease and tumor shrinkage (40 mm × 40 mm × 35 mm, 32 mm × 25 mm × 31 mm, respectively) with reduced right optic nerve invasion. Bromocriptine was replaced with cabergoline (a maximum dose of 4 mg/week), resulting in remarkable reduction of headaches.
Due to the young age of the patient at the onset of the disease, genetic testing for AIP and MEN1 mutations was carried out and yielded negative results. Echocardiography was performed as part of screening for cabergoline‑induced valvulopathy in patients on high‑dose therapy (>2 mg/week), and no abnormalities were detected.
The tumor dimensions have been stable since December 2019 (25 mm × 13 mm × 23 mm), with an increase in the cystic vs solid structure of the tumor (Figure 1C–1G). The patient is asymptomatic and continues the cabergoline treatment (1.5 mg/week). Prolactin levels remain increased (4500 μlU/ml on average; 20% of the baseline levels).
GPs usually respond well to dopamine agonists (DAs), and cabergoline is the first‑line treatment due to its higher efficacy as compared with bromocriptine.2 Tumor volume reduction by at least 25% and normoprolactinemia at 3 months after DA treatment initiation are associated with a high probability of achieving a long‑term complete response.3
Surgical management of GP is indicated in pituitary apoplexy, cerebrospinal fluid rhinorrhea, and mass growth progression despite optimal treatment, that is, in tumors resistant to DA.2,4 In aggressive prolactinomas, temozolomide or pasireotide can be a part of multimodal treatment.2,5
Our case shows that a conservative pharmacologic approach to GP could be a first‑line treatment, even in the case of incomplete response to cabergoline. Considering the risk of surgery, partial reduction of the tumor and cystic degeneration after DA therapy, as well as significant decrease in serum prolactin levels (albeit non‑normalized) and remission of headaches, the treatment should be considered partially successful.
- Maiter D, Delgrange E. Therapy of endocrine disease: the challenges in managing giant prolactinomas. Eur J Endocrinol. 2014; 170: R213‑R227. | Crossref
- Petersenn S, Fleseriu M, Casanueva FF, et al. Diagnosis and management of prolactin‑secreting pituitary adenomas: a Pituitary Society International Consensus Statement. Nat Rev Endocrinol. 2023; 19: 722‑740. | Crossref
- Lee Y, Ku CR, Kim EH, et al. Early prediction of long‑term response to cabergoline in patients with macroprolactinomas. Endocrinol Metab (Seoul). 2014; 29: 280‑292. | Crossref
- Zielinski G, Ozdarski M, Maksymowicz M, et al. Prolactinomas: prognostic factors of early remission after transsphenoidal surgery. Front Endocrinol (Lausanne). 2020; 11: 439. | Crossref
- Coopmans EC, van Meyel SWF, Pieterman KJ, et al. Excellent response to pasireotide therapy in an aggressive and dopamine‑resistant prolactinoma. Eur J Endocrinol. 2019; 181: K21‑K27. | Crossref
SUPPLEMENTARY MATERIAL
ARTICLE INFORMATION