Complete remission of hypercortisolemia in a patient with severe ectopic Cushing syndrome caused by small-cell lung cancer

 CC BY 4.0
CC BY 4.0
Complete remission of hypercortisolemia in a patient with severe ectopic Cushing syndrome caused by small-cell lung cancer
Ectopic Cushing syndrome (ECS) accounts for 10%–20% of all CS cases. The second most common cause of ECS is small‑cell lung cancer (SCLC).1,2 However, recent reports indicate that CS in the course of SCLC might be underestimated as hypercortisolemia and often remains undiagnosed.2,3 ECS in SCLC is often associated with severe hypercortisolemia and poor prognosis.3 We report for the first time a case of complete remission of hypercortisolemia in ECS caused by SCLC.
A 65‑year‑old man was admitted to a hospital due to hypertension (220/120 mm Hg), lower limb edema, and severe hypokalemia (2.16 mmol/l; reference range [RR], 3.5–5.1 mmol/l), lasting for about a month. Physical examination showed subtle body changes, such as moon face, redistribution of fat tissue, and plethora. Hormonal assessment suggested ECS—no inhibition in the low- and high‑dose dexamethasone test, high adrenocorticotropic hormone levels (222 pg/ml; RR, 6–56 pg/ml). Computed tomography (CT) showed 2 polycyclic lesions in the inferior lobe of the left lung and metastatic mediastinal and abdominal lymph nodes (Figure 1A). Histopathologic examination of the bronchoscopic biopsy specimen showed SCLC. Treatment with metyrapone was implemented with an increasing dose of up to 3250 mg per day. In the sixth week of hypercortisolemia treatment, a palliative chemoimmunotherapy with a carboplatin, etoposide, and atezolizumab combination was initiated, followed by prophylactic cranial radiotherapy. The chemoimmunotherapy regimen underwent only minor changes during the treatment period (Figure 1B). After 2 months of treatment (following the second cycle of chemoimmunotherapy), metyrapone was changed to osilodrostat with a starting dose of 4 mg per day, later titrated based on urine free cortisol (UFC) values (Figure 1C and 1D). Shortly after decreasing the dose of osilodrostat to 1 mg per day, dexamethasone was introduced due to immunotherapy‑related autoimmune hepatitis (Figure 1C and 1D). During treatment, apart from biochemical improvement (based on UFC level [Figure 1C] and normal circadian cortisol rhythm with midnight cortisol levels of 3.32 µg/dl), imaging showed a partial response of SCLC (regression of the tumor mass on control CT [Figure 1E]). Furthermore, a significant clinical improvement was noted, manifested as regression of peripheral edema, plethora, and moon face, better control of hypertension and hypokalemia, and reduction of potassium supplementation and antihypertensive drugs. According to the Response Evaluation Criteria In Solid Tumors 1.1 guidelines,4 partial response was observed. After treatment, the main tumor decreased from 52 mm to 16 mm. Due to the complete remission of CS (morning cortisol levels at 6 AM, 77 µg/dl), the osilodrostat treatment was discontinued after 13 months. Adrenal insufficiency was excluded, as the stimulation test with 1 μg of tetracosactide yielded normal cortisol output. Control biochemical tests performed 1 month after osilodrostat discontinuation showed eucortisolemia. Recently, the patient has received the 27th cycle of maintenance immunotherapy. Nearly 2 years after the diagnosis, he remains clinically stable with no symptoms of CS.
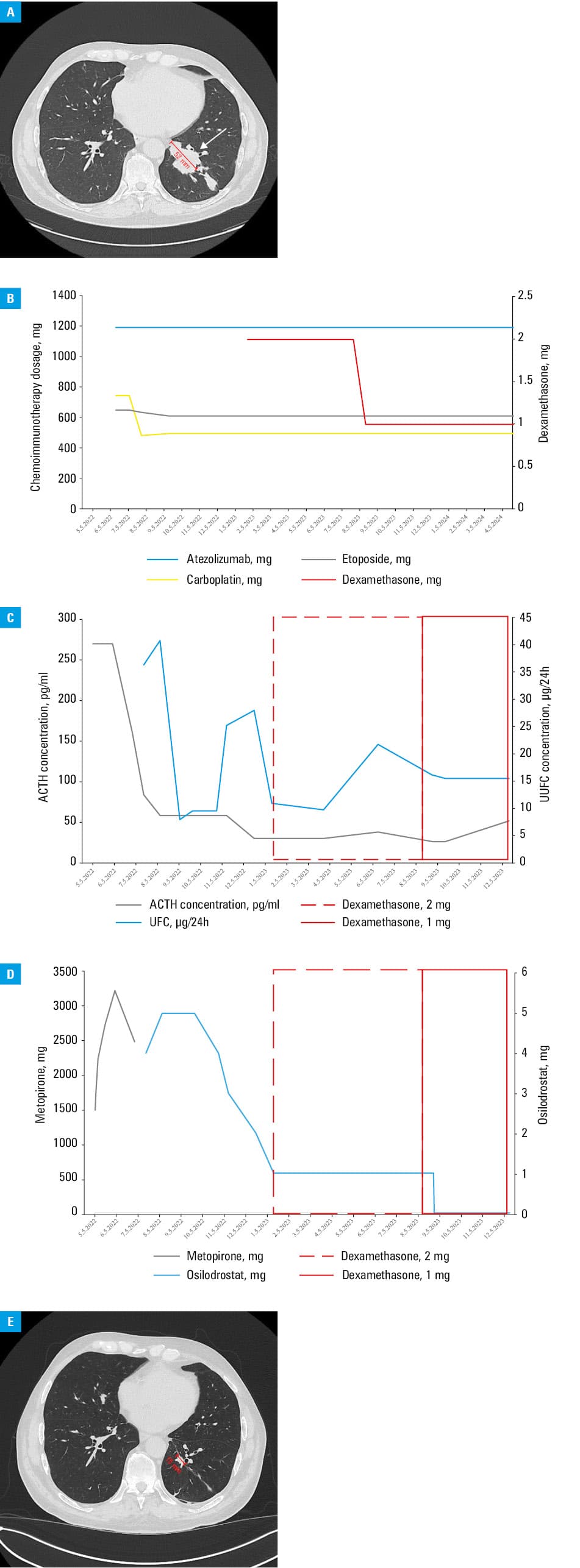
Our case shows that immediate and effective treatment of life‑threatening hypercortisolemia, leading to a diagnosis and rapid initiation of chemoimmunotherapy, improves prognosis and may result in long‑term remission. It also confirms that osilodrostat is an effective and easy‑to‑titrate drug in treating hypercortisolemia in ECS.5,6
- Ragnarsson O, Juhlin CC, Torpy DJ, Falhammar H. A clinical perspective on ectopic Cushing’s syndrome. Trends Endocrinol Metab. 2023; 23: 1043‑1057.
- Young J, Haissaguerre M, Viera‑Pinto O, et al. Management of endocrine disease: Cushing’s syndrome due to ectopic ACTH secretion: an expert operational opinion. Eur J Endocrinol. 2020; 182: 29‑58. | Crossref
- Piasecka M, Larsson M, Papakokkinou E, et al. Is ectopic Cushing’s syndrome underdiagnosed in patients with small cell lung cancer? Front Med (Lausanne). 2022; 30: 95‑102. | Crossref
- Eisenhauer EA, Therasse P, Bogaerts J et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009; 45: 228‑247. | Crossref
- Dormoy A, Haissaguerre M, Vitellius G, et al. Efficacy and safety of osilodrostat in paraneoplastic Cushing syndrome: a real‑world multicenter study in France. J Clin Endocrinol Metab. 2023; 108: 1475‑1487. | Crossref
SUPPLEMENTARY MATERIAL
ARTICLE INFORMATION