A case of systemic lupus erythematosus with antineutrophil cytoplasmic antibodies–positive skin vasculitis and concomitant combined thrombophilia
1,2

 CC BY 4.0
CC BY 4.0
A case of systemic lupus erythematosus with antineutrophil cytoplasmic antibodies–positive skin vasculitis and concomitant combined thrombophilia
We present a case of a 31‑year‑old man diagnosed with systemic lupus erythematosus (SLE) and antiphospholipid syndrome (APS). His medical history included fever, malar rash, arthritis, pulmonary embolism, and deep vein thrombosis. He had elevated antinuclear antibodies (ANA) titer (1:640; reference range [RR] <1:160), positive anticardiolipin antibodies in immunoglobulin G (IgG) and immunoglobulin M (IgM) classes, and anti–B2‑glucoprotein I antibodies in IgM class antibodies, confirmed in subsequent tests (Supplementary material, Table S1). Initial treatment with systemic glucocorticoids (SGCs) resulted in clinical improvement.
Four years later, in 2013, the patient was admitted to the Department of Allergy and Immunology at the University Hospital in Kraków with SLE exacerbation. However, subsequent SLE exacerbations were accompanied by skin vasculitis. Laboratory workup showed high ANA titers (1:>20 480), positive anti–double‑stranded DNA (anti‑dsDNA) antibodies confirmed by Crithidia luciliae indirect immunofluorescence test at a titer of 1:320 (RR <1:10), leucopenia, lymphopenia, decreased complement components C3 and C4 levels, proteinuria, and granular casts in the urine sediment (Supplementary material, Table S1). SGCs were administered, leading to SLE remission. Given the history of thromboembolic events, inherited thrombophilias were also evaluated, showing reduced activity of total protein S (26%; RR, 60%–130%) and heterozygous factor V Leiden gene mutation.
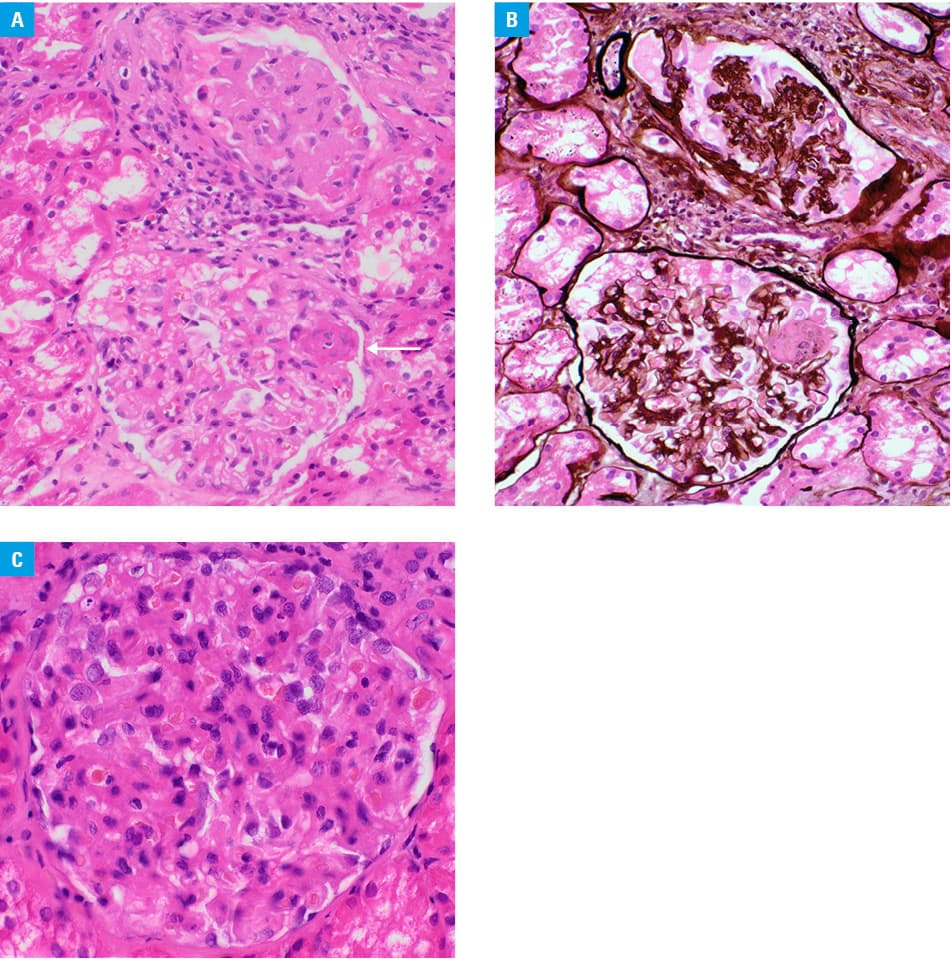
One year later, the patient was hospitalized again due to another SLE exacerbation, this time overlapping with new‑onset skin vasculitis. He presented with necrotic skin ulcerations on both legs. Laboratory results revealed leucopenia, lymphopenia, decreased complement components C3 and C4, proteinuria, erythrocyturia, and granular casts in the urine sediment (Supplementary material, Table S1). Immunological tests confirmed ANA at a titer of above 1:20 480 and identified anti‑dsDNA, anti‑nucleosome, anti–Sjögren‑syndrome‑related antigen A, antihistone, and anti–Ro52 antibodies (Supplementary material, Table S1). Furthermore, a high titer of antineutrophil cytoplasmic antibodies (ANCA; 1:40; RR <1:10) directed against proteinase‑3 (PR3; >200 RU/ml; RR <20 RU/ml) was detected (Supplementary material, Table S1). A kidney biopsy revealed class IV lupus nephritis according to the 2018 International Society of Nephrology / Renal Pathology Society (ISN / RPS) pathological classification (Figure 1A–1C).1 Before being processed in the routine manner, the biopsy was divided into portions for optical microscopy, fluorescence microscopy, and electron microscopy. The part processed for optical microscopy contained 11 glomeruli. They showed increase in mesangial matrix and hypercellularity. The capillary walls were significantly thickened. One glomerulus contained a segmental area of fibrinoid necrosis. There were foci of interstitial fibrosis with concomitant tubular atrophy. The extraglomerular vessels present on the biopsy showed some ateriosclerosis but no vasculitis. On electron microscopy, subendothelial, intramembranous, and mesangial electron‑dense deposits were seen together with mesangial interposition. Immunofluorescence was mostly negative (in the view of EM findings, this could be interpreted as false negative). The entire picture was compatible with mesangioproliferative pattern of damage and was classified as IV G (Figure 1A–1C) in concordance with clinical data according to the ISN / RPS system.1 Treatment with SGCs and cyclophosphamide (total dose of 12 g) was initiated, but the patient did not achieve full remission.
In 2015, persistent left leg ulceration, leucopenia, lymphopenia, decreased complement component C3, positive anti‑dsDNA (titer 1:2 560), and anti–PR3 antibodies (86.8 RU/ml) led to treatment with rituximab (total dose 2 g; Supplementary material, Table S1).
Then, the patient presented with severe complications, including extensive infected ulcers with necrosis on the right lower limb (requiring amputation), sacral pressure ulcer, septic shock, acute kidney failure, multiple cardiac arrests due to ventricular fibrillation with successful resuscitations, and infective endocarditis complicated by implantation of a cardioverter‑defibrillator.
During 9‑year follow‑up, the patient experienced 2 more SLE exacerbations requiring hospitalization—in 2018, when cyclosporine was added to the treatment, and in 2022, when cyclosporine was switched to methotrexate. Currently, the patient is under constant monitoring at the Immunology Outpatient Clinic of the University Hospital in Kraków. He is in stable condition and is treated with methylprednisolone (4 mg/day) and methotrexate (10 mg/week).
SLE and ANCA‑associated vasculitis (AAV) are severe autoimmune diseases that share some clinical features, such as cutaneous lesions, arthritis, or renal involvement.2,3 In the majority of patients, SLE and AAV can be distinguished by demographic characteristics or laboratory and histopathology results, while in some cases they display mixed patterns matching the classification criteria of both diseases.3 Using enzyme‑linked immunosorbent assay, Galeazzi et al4 underlined that in 9.3% of SLE cases, the patient suffered from antimyeloperoxidase positive AAV, whereas only up to 1.7% of patients had anti‑PR3 positive AAV.4 More than 90% of SLE‑AAV cases present with serious manifestations, for example, glomerulonephritis, and have to be treated aggressively with glucocorticoids and cyclophosphamide.2 The coexistence of SLE and AAV might be associated with a high mortality rate of 13%.2 Additionally, our case shows how important it is to perform a diagnosis toward APS, but also to extend diagnostics with testing for inherited thrombophilias.5 Mortality in SLE patients is higher than in healthy controls, and is primarily a consequence of high SLE activity, infections, and cardiovascular complications.6
- Bajema IM, Wilhelmus S, Alpers CE, et al. Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: clarification of definitions, and modified National Institutes of Health activity and chronicity indices. Kidney Int. 2018; 93: 789‑796. | Crossref
- Itikyala S, Pattanaik D, Raza S. Systemic lupus erythematosus (SLE) and antineutrophil cytoplasmic antibody‑associated vasculitis (AAV) overlap syndrome: case report and review of the literature. Case Rep Rheumatol. 2019; 2 019: 5013904. | Crossref
- Jarrot PA, Chiche L, Hervier B, et al. Systemic lupus erythematosus and antineutrophil cytoplasmic antibody‑associated vasculitis overlap syndrome in patients with biopsy‑proven glomerulonephritis. Medicine (Baltimore). 2016; 95: e3748. | Crossref
- Galeazzi M, Morozzi G, Sebastiani GD, et al. Anti‑neutrophil cytoplasmic antibodies in 566 European patients with systemic lupus erythematosus: prevalence, clinical associations and correlation with other autoantibodies. European Concerted Action on the Immunogenetics of SLE. Clin Exp Rheumatol. 1998; 16: 541‑546.
- Linnemann B, Hart C. Laboratory diagnostics in thrombophilia. Hamostaseologie. 2019; 39: 49‑61. | Crossref
SUPPLEMENTARY MATERIAL
ARTICLE INFORMATION