Erdheim–Chester disease (ECD) is a rare entity belonging to the “L” group of Langerhans histiocytoses.1 To date, fewer than 1000 cases of ECD have been described in the literature. The disease is more common in men (72%), and the median age at diagnosis is 56 years.2,3 Bone pain, constitutional symptoms, xanthelasma, ataxia, ocular exophthalmos, and tamponade may indicate the onset of the disease, and their frequency varies considerably between patients. The most common initial symptom, uremia of undetermined origin (16%), can occur many years before the diagnosis of ECD.3
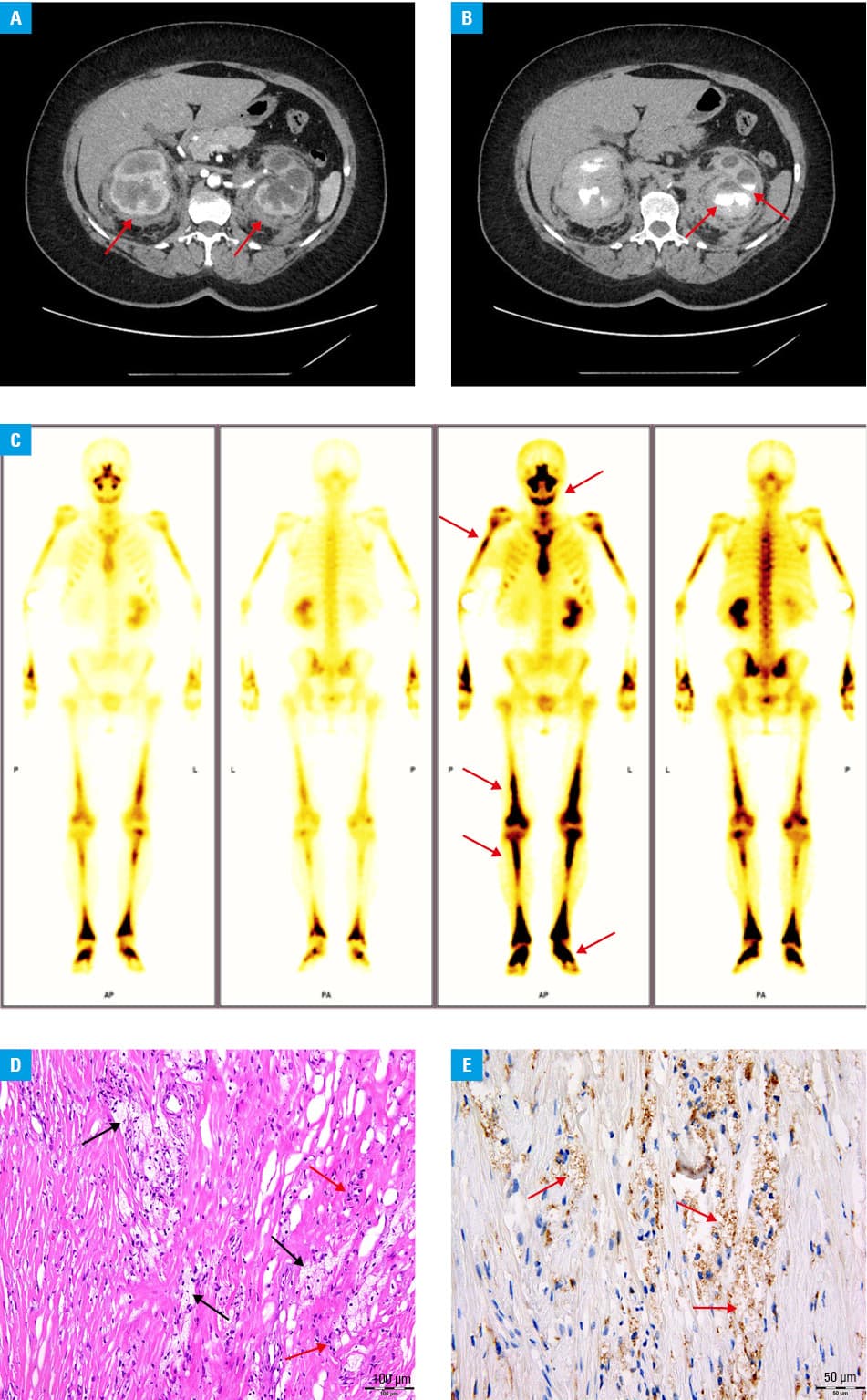
We present a case of a 55‑year‑old woman who was referred to a nephrology department because of recurrent complicated cystitis, bilateral pain in the lumbar region and perinephric lesions found on computed tomography (CT). Several months earlier, she had been diagnosed with myocarditis; however, endomyocardial biopsy had not been performed. On admission, the patient had symptoms of acute complicated urinary tract infection with fever, vomiting, abdominal tenderness, elevated C‑reactive protein level (163 mg/l; reference range [RR] <5 mg/l) and white blood cell count (18.44 × 109/l; RR, 4–10 × 109/l), leukocyturia, and bacteriuria; therefore, an empiric broad‑spectrum antibiotic was administered. Blood and urine cultures showed Escherichia coli infection, and the treatment was changed to targeted antibiotic therapy. After recovery, a comparative abdominal and pelvic CT scan showed progression of the perinephric infiltrates, demonstrating a “hairy kidney” sign (Figure 1A), which is considered pathognomonic for ECD. Additionally, thickening of the soft tissues of the ureter causing hydronephrosis (Figure 1B), and osteosclerotic lesions in the femurs were visualized. Whole‑body scintigraphy using 99mtechnetium‑methylene diphosphonate showed bilateral diffuse increased tracer uptake in the proximal humeral epiphysis and shaft and in the distal femoral epiphysis (Figure 1C). To confirm the diagnosis, laparoscopy with biopsy of the perirenal mass was performed. The biopsy showed a histiocyte infiltrate with foam‑like cytoplasm on hematoxylin and eosin staining (Figure 1D), and histiocytes positive for CD68 (Figure 1E) and negative for CD1, S100, and langerin. Such a histiologic picture differs from that observed in other types of histiocytic neoplasms, such as Langerhans cell histiocytosis and Rosai–Dorfman disease. The BRAFV600E mutation was confirmed using the Cobas 4800 BRAFV600 Mutation Test kit (Roche Diagnostics, Basel, Switzerland). Based on the correlation of clinical features with radiological and histological findings, a diagnosis of ECD was made. Targeted therapy with vemurafenib at a dose of 480 mg orally twice daily was started, with minor side effects (folliculitis, alopecia, and skin rash) acceptable to the patient. After 3 months of BRAF inhibitor therapy, follow‑up abdominal and pelvic CT scans showed no radiographic progression of the disease, and no urinary tract infections have been observed since the treatment initiation.
Patients with ECD present with a variety of nonspecific symptoms, often with multiorgan manifestations.4 Involvement of the central nervous system, retroperitoneal space, and lungs is associated with worse survival rates. Cohort studies showed an overall mortality rate of 24.8% in ECD, with a median survival time of 162 months and a 5‑year survival rate of 82.7%.3 In addition to conventional therapies (corticosteroids, interferon-α, chemotherapy), targeted therapies with BRAF inhibitors, MEK inhibitors, or combinations thereof have shown promising results.5 Due to its complexity, diagnosis and treatment of ECD are challenging for physicians, and further research into innovative diagnostic and treatment options is warranted.
- Emile J‑F, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage‑dendritic cell lineages. Blood. 2016; 127: 2672‑2681. | Crossref
- Haroche J, Cohen‑Aubart F, Amoura Z. Erdheim‑Chester disease. Blood. 2020; 135: 1311‑1318. | Crossref
- Cohen‑Aubart F, Emile J‑F, Carrat F, et al. Phenotypes and survival in Erdheim‑Chester disease: results from a 165‑patient cohort. Am J Hematol. 2018; 93: E114‑E117. | Crossref
- Verschelden G, Van Laethem J, Velkeniers B, et al. Significant response to dabrafenib in a patient with Erdheim‑Chester disease with BRAFV600E mutation. Pol Arch Intern Med. 2018; 128: 386‑388. | Crossref
- Goyal G, Heaney ML, Collin M, et al. Erdheim‑Chester disease: consensus recommendations for evaluation, diagnosis, and treatment in the molecular era. Blood. 2020; 135: 1929‑1945. | Crossref
ARTICLE INFORMATION