A definition of pyrexia of unknown origin (PUO) has evolved over the years but the diagnostic challenges remain the same. Most recently, PUO investigations have been subcategorized into classical, nosocomial, HIV‑related, and neutropenic. Broadly speaking, the etiologies fall under one of the categories encompassing infection, inflammation, neoplastic process, or miscellaneous.
We present a case of PUO in a 68‑year‑old, fit, and healthy man that illustrates the importance of a methodical approach and lateral thinking. The patient was of Indian origin, and immigrated to the United Kingdom in 1958. He presented with a 6‑month history of fever, night sweats, and fatigue. He had a 4‑day history of bilateral leg and testicular swelling, as well as decreased oral intake. The patient had a tuberculosis (TB) contact history, as his mother was diagnosed with and treated for tuberculosis in 1964. No other symptoms including weight loss or no significant past medical conditions were reported. On examination, he was febrile at 38 °C with blood pressure of 90/70 mm Hg and no obvious lymphadenopathy. There was bilateral pitting edema up to his groin with scrotal sac swelling without obvious testicular or penile anomalies, as confirmed on ultrasound. Blood counts on admission, as compared with normal results a month ago, showed anemia (hemoglobin, 111 g/l; reference range [RR], 115–165 g/l), thrombocytopenia (91 × 109/l; RR, 140–400 × 109/l), and mild elevation of C‑reactive protein (CRP; 40 mg/l; RR, 0–10 mg/l). Other parameters were normal. Chest X‑ray was unremarkable. Given the contact history of exposure and his ethnic background, he was investigated for presumed TB.
Computed tomography (CT) of the chest and abdomen showed thickening of the upper esophagus with dysmorphic calcification, multiple mural calcifications in the small bowel with terminal ileal thickening, and multiple abnormal ileocolic nodes. Gastroscopy demonstrated a chronic Helicobacter pylori infection and eradication therapy was initiated. Twelve biopsies from different sites were taken. Colonoscopy showed normal architecture and 10 biopsies were taken. They were negative for Whipple’s disease, TB, and cytomegalovirus (CMV). Blood‑borne viral screening, Epstein‑Barr virus (EBV), and CMV assays were negative. In addition, 5 blood cultures, 2 malaria assays, 2 urine cultures, 3 stool cultures, amebiasis serology, 3 sputum samples (including TB culture and TB polymerase chain reaction), 2 respiratory viral screenings, and 9 COVID‑19 assays were negative. Transthoracic echocardiogram confirmed normal cardiac function, with no vegetations or thrombi. Subsequent blood tests showed worsening thrombocytopenia (79 × 109/l), anemia (hemoglobin, 90 g/l), worsening coagulopathy (international normalized ratio, 1.2; prothrombin time, 16.2 s; RR, 12–15 s, activated partial thromboplastin time [APTT], 62.9 s; RR, 26.1–36.6 s, fibrinogen, 3.7 g/l; RR, 2–4 g/l), and elevated lactate dehydrogenase (LDH) of 1279 U/l (RR, 150–246 U/l). Blood film showed normal platelet and red blood cell (RBC) morphology. The Coombs test was negative. Another CT showed an improvement in the thickening noted in the esophagus, small bowel, and a decrease in the size of the ileocolic nodes.
Two months into his admission, the patient remained febrile at 39 °C, had consistently low blood pressure, which eventually became refractory to fluid resuscitation while on TB treatment, meropenem, and vancomycin. He was moved to intensive care unit (ICU) for inotropic support and got further 2 CT scans, which showed mild splenomegaly. Further workup for PUO was undertaken. Filarial serology, syphilis assay, yersinia, leptospirosis, schistosomiasis, strongyloides assay, antistreptolysin O titer, 3 malaria assays, human herpesvirus (HHV) 6, HHV7, and HHV8 assays were negative. Further 3 blood cultures were also negative. Positron emission tomography / CT (PET/CT) was requested. Liver function test revealed gradual decline in total albumin from normal to 20 g/l (RR, 35–50 g/l), likely reflecting a negative‑phase reaction. Edema progressed to the abdominal wall, sacrum, and bilateral arms. The patient complained of thigh pain and swelling. On examination, this was extremely difficult to ascertain, but raised the possibility of a brawny swelling with ill‑defined margins and magnetic resonance imaging (MRI) of the thigh was requested to investigate the swelling.
Two extensive autoimmune screenings along with myositis and myeloma assays were negative. Hemophagocytic lymphohistiocytosis (HLH) was considered as a diagnosis, given the unexplained biochemical decline, and worsening clinical picture, but the HLH probability score was 5%. Plasma viscosity was elevated, ferritin level was 1477 µg/l (RR, 22–322 µg/l), LDH increased to 2931 IU/l, triglycerides to 3.55 mmol/l (RR, 0–1.7 mmol/l), aspartate aminotransferase to 82 IU/l (RR, 8–33 IU/l), and bone marrow biopsy showed no macrophage activity. A small B‑cell clone was identified on flow cytometry, but its significance was unclear. All cell lines appeared normal. At ICU the patient was initiated on 50 mg hydrocortisone every 6 hours to treat sepsis and possible inflammatory pathologies, and he improved dramatically. Discussions with rheumatology and hematology departments ensued with a consensus that PET‑CT would likely detect the underlying pathology, which both felt was likely an infection. Further blood tests showed worsening thrombocytopenia, sustained with frequent platelet transfusions, worsening coagulation, and decline in hemoglobin to 80 g/l. TB treatment was held due to bicytopenia, while the other antibiotics were continued.
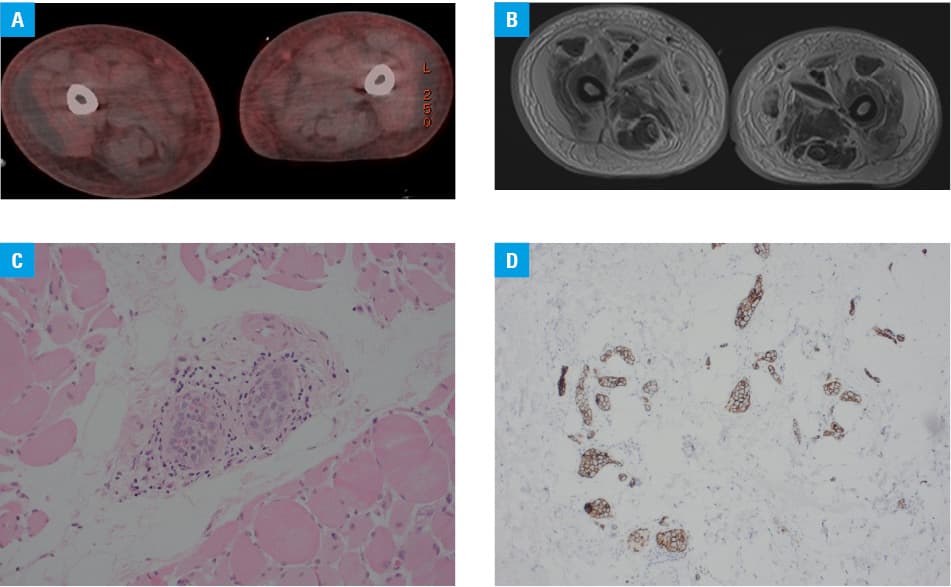
The PET/CT scan showed generalized edema with mild hepatosplenomegaly, without other abnormalities (Figure 1A). The patient started deteriorating on steroid treatment. Two and a half months after the admission, the patient had an extensive infection screening including 8 blood cultures, 2 extensive autoimmune screenings, 1 bone marrow biopsy, 6 CT scans, 8 chest X‑rays, 2 echocardiographies, and 1 PET/CT; all leading to a giant question mark. The patient was deteriorating with skin turning ash‑grey. EBV and CMV assays were repeated and showed EBV reactivation with 14 000 DNA copies. Finally, MRI of the thigh showed widespread edema in both thighs with layers of fluid tracking along the muscle, with suspected muscle necrosis, and small abscesses without obvious gangrene (Figure 1B). A biopsy of the muscle was taken. While waiting for the biopsy results, the patient developed refractory thrombocytopenia. Due to bicytopenia, reactivation of EBV, and hepatosplenomegaly, the possibility of HLH was raised again. Another bone marrow biopsy showed macrophages with hemophagocytosis of mature RBC in the bone marrow. Our suspicion of HLH was confirmed. Methylprednisolone was given as part of the first‑line management for HLH. The muscle biopsy confirmed intravascular lymphoma (IVL; Figure 1C and 1D). Unfortunately, the patient passed away shortly after chemotherapy was commenced.
HLH is a life‑threatening hyperinflammatory syndrome secondary to uncontrolled activation of T‑cells, NK cells, and macrophages leading to immune‑mediated injury in multiple organs.1 Secondary HLH is caused by infection, autoimmune disease, or malignancy. Initially, the HLH‑2004 protocol was used to treat HLH but due to its limitations, it was replaced with the H‑score, which incorporates many parameters, including immunodeficiency. Adverse prognostic factors are male sex, APTT above 36 seconds, LDH above 1000 U/l, and CRP above 100 mg/l.2 The first‑line treatment is high‑dose corticosteroid, such as methylprednisolone alone or in combination with anakinra.2 Other treatment options are polyvalent immunoglobulins, etoposide, and anticytokine modulators.1,3
IVL is a rare, elusive, and clinically aggressive lymphoma of the large cells. In the past, the diagnosis was usually made during an autopsy. The cells are immunophenotypically similar to mature B‑cells.4 There are 3 variants of this pathology, namely classical, cutaneous, and HLH. The latter is very aggressive, with a survival time of 2–8 months.5 There is no standardized staging unique to IVL, but the Ann Arbor staging can be used. The treatment regime is R‑CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone). Ferreri et al4 concluded that presentation of IVL in Asian and western population varies, with a better prognosis noted in the Western population. In the western countries, R‑CHOP had a better outcome, with 91% overall response and 88% complete remission, with fewer favorable outcomes for the Japanese counterparts.5
In conclusion, this case highlights 3 main learning points. Firstly, the importance of detailed history taking and clinical examination. Secondly, clinical suspicion of HLH, due to the wide range of its triggers and high mortality, can help with diagnosis and early management. Finally, the importance of perseverance and to always pull at any loose threads.
- Bichon A, Bourenne J, Allardet‑Serven J, et al. High mortality of HLH in ICU regardless etiology or treatment. Front Med (Lausanne). 2021: 8: 735796. | Crossref
- Zhang Q, Lin Y, Bao Y, et al. Analysis of prognostic risk factors and establishment of prognostic scoring system for secondary adult hemophagocytic syndrome. Curr Oncol. 2022; 29: 1136‑1149. | Crossref
- Mehta P, Cron RQ, Hartwell J, et al. Silencing the cytokine storm: the use of intravenous anakinra in hemophagocytic lymphohistiocytosis or macrophage activation syndrome. Lancet Rheumatol. 2020; 2: e358‑e367. | Crossref
- Ferreri AJM, Dognini GP, Campo E, et al. Variations in clinical presentation, frequency of hemophagocytosis and clinical behavior of intravascular lymphoma diagnosed in different geographical regions. Hematologica. 2007; 92: 486‑492. | Crossref
- Ponzoni M, Campo E, Nakamura S. Intravascular large B‑cell lymphoma: a chameleon with multiple faces and many masks. Blood. 2018; 132: 1561‑1567. | Crossref
ARTICLE INFORMATION