Chronic Budd–Chiari syndrome as an initial manifestation of primary antiphospholipid syndrome in a young patient



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Chronic Budd–Chiari syndrome as an initial manifestation of primary antiphospholipid syndrome in a young patient
Budd–Chiari syndrome (BCS) is a liver dysfunction caused by obstruction of hepatic venous outflow.1 The most frequent underlying causes of BCS are myeloproliferative syndromes and hypercoagulable states that occur in certain cancers, infections, and autoimmune diseases.2 The association with antiphospholipid syndrome has been described before; however, it requires high clinical suspicion,3 especially in the absence of previous thromboembolic events and due to a wide spectrum of possible presentations.4,5
A previously healthy 18‑year‑old man presented with a 1.5‑year history of progressive ascites and collateral periumbilical venous circulation extending to the thorax (Figure 1A). He also had nonpainful erythematous papules on the fingers of both hands (Figure 1B), palmo‑plantar erythema, and stasis dermatitis with a symmetrical distribution on the lower extremities. The initial laboratory workup showed a hemoglobin level of 11.9 g/dl (reference range [RR], 13–17 g/dl) and platelet count of 115 000/µl (RR, 140 000–450 000/µl). The coagulation profile was prothrombin time, 58% (RR, 70%–110%); activated partial thromboplastin time, 57 s (RR, 30–45 s); total bilirubin, 2.3 mg/dl (RR, 0.4–1.4 mg/dl); direct bilirubin, 1.2 mg/dl (RR, 0.1–0.4 mg/dl); with slightly increased levels of liver enzymes and serum albumin level of 4.4 g/dl (RR, 3.2–5 g/dl). Ascitic fluid analysis showed a leukocyte count of 300 cells/µl, protein level of 5.2 g/dl, and negative cultures for common microorganisms. Serum ascitic albumin level gradient was 0.8. Cytology of the ascitic fluid was positive for lymphocytosis, but no malignant cells were identified. Test results for hepatotropic viruses and markers of hepatic storage diseases, such as Wilson disease and hemochromatosis, were negative. In addition, antibodies for autoimmune hepatitis were absent and the level of α1‑antitrypsin was within the reference range.
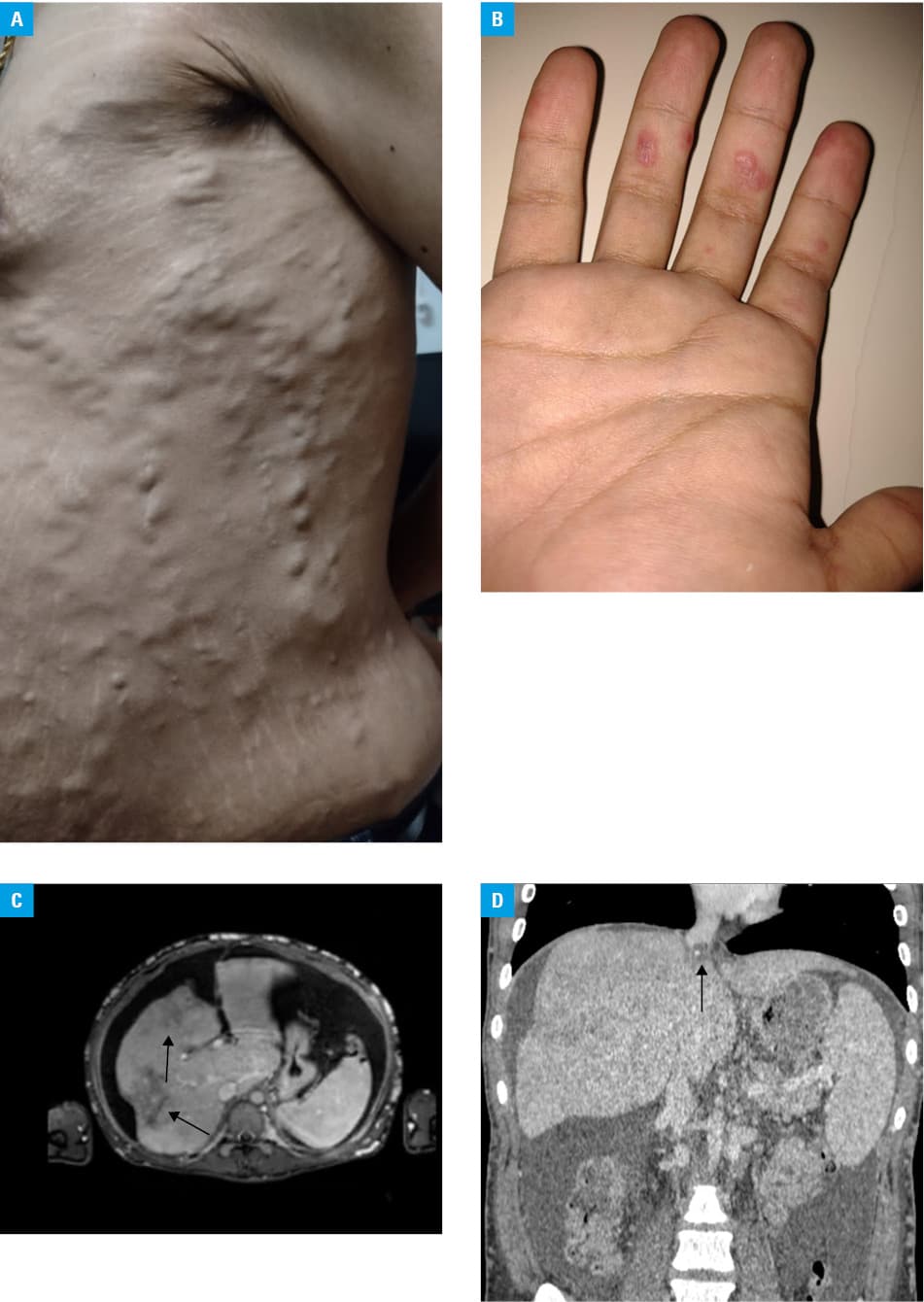
Anticardiolipin immunoglobulin G antibodies, lupus anticoagulant, and anti-β2 glycoproteins were present (triple positive), which lead to a diagnosis of primary antiphospholipid syndrome not associated with systemic lupus erythematosus, as determined by a negative result of the antinuclear antibody test. Imaging studies were performed, including splenoportal Doppler ultrasonography, computed tomography angiography, and magnetic resonance imaging of the abdomen, which showed alteration of hepatic perfusion, without portal thrombosis and with hepatopetal flow (Figure 1C). Transjugular hepatic venous pressure measurement was also carried out, indicating an increased portosystemic gradient (10 mm Hg) and severe stenosis of the suprahepatic veins secondary to the presence of thrombus at this level (Figure 1D). Based on these findings, a diagnosis of BCS was made. No esophageal varices were found on video‑assisted endoscopy of the upper digestive tract, and transesophageal Doppler echocardiography showed moderate mitral regurgitation linked to the antiphospholipid syndrome.
This case describes an atypical manifestation of primary antiphospholipid syndrome in a young patient with no history of thrombosis, who initially presented with slow‑onset portal hypertension secondary to BCS and eventually developed liver cirrhosis due to a delayed diagnosis. BCS is a rare disorder that is difficult to diagnose, especially in a chronic course. Identifying the cause is always challenging, and timely diagnosis is crucial to prevent severe comorbidities.
- Sharma A, Keshava SN, Eapen A, et al. An update on the management of Budd‑Chiari syndrome. Dig Dis Sci. 2021; 66: 1780‑1790. | Crossref
- Solela G, Daba M. Budd‑Chiari syndrome as an initial presentation of systemic lupus erythematosus associated with antiphospholipid syndrome: a case report with review of the literature. Open Access Rheumatol. 2023; 15: 139‑143. | Crossref
- Uthman I, Khamashta M. The abdominal manifestations of the antiphospholipid syndrome. Rheumatology (Oxford). 2007; 46: 1641‑1647. | Crossref
- Martens P, Nevens F. Budd‐Chiari syndrome. United European Gastroenterol J. 2015; 3: 489‑500. | Crossref
- Chinen N, Koyama Y, Sato S, Suzuki Y. A case of acute Budd‑Chiari syndrome complicating primary antiphospholipid syndrome presenting as acute abdomen and responding to tight anticoagulant therapy. Case Rep Rheumatol. 2016; 2016: 1‑5. | Crossref
ARTICLE INFORMATION