Glu112Lys: a rare pathogenic transthyretin gene variant associated with negative scintigraphy findings
1,2,3 ,
,

 CC BY 4.0
CC BY 4.0
Glu112Lys: a rare pathogenic transthyretin gene variant associated with negative scintigraphy findings
Hereditary transthyretin amyloidosis (ATTRm), an autosomal dominant disorder, encompasses a broad spectrum of pathogenic transthyretin (TTR) gene variants, with over 100 identified to date.1 Moreover, ATTRm, once considered a rare disease beyond endemic regions, has become more readily identified due to the application of optimized and simplified diagnostic algorithms, including scintigraphy with 99mtechnetium (99mTc) and bone‑avid tracers.2,3 Importantly, the degree of tracer uptake may vary depending on the subtypes of amyloid fibrils.4
A 61‑year‑old man with heart failure and unexplained cardiac hypertrophy presented with echocardiographic findings of concentric left ventricular (LV) hypertrophy (LV mass, 400 g), decreased LV ejection fraction (34%), and decreased global LV longitudinal strain (–10.8%) exhibiting an “apical sparing” pattern and third‑degree diastolic dysfunction (Figure 1A and 1B). Scintigraphy with 99mTc‑3,3‑diphosphono‑1,2‑propanodicarboxylic acid ([99mTc]Tc‑DPD) did not show a significantly increased myocardial uptake (Figure 1C). A biopsy confirmed deposits of amyloid and TTR protein (Figure 1D and 1E). Genetic testing revealed the presence of a rare pathological variant of the TTR gene, namely Glu112Lys. The patient was diagnosed with ATTRm cardiomyopathy and scheduled for pharmacological treatment with tafamidis. Scintigraphy was repeated several months after the initial negative test results, and the findings remained negative. Evaluation of family members confirmed the presence of the Glu112Lys TTR gene variant in 2 asymptomatic daughters.
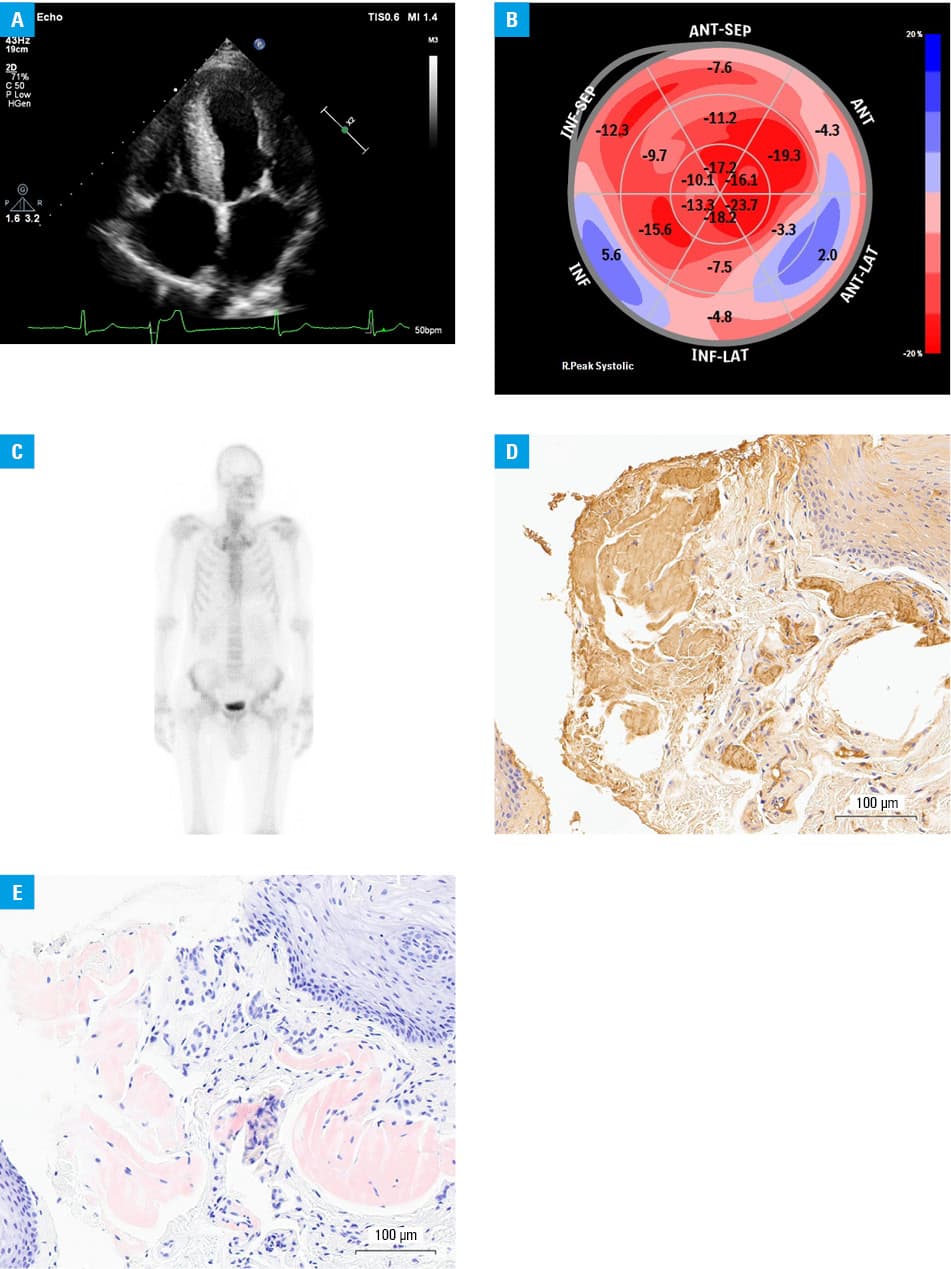
Scintigraphy with 99mTc has emerged as a valuable diagnostic tool for assessing cardiac involvement in ATTRm. This imaging modality employs bone‑avid tracers to visualize amyloid deposits in the heart, providing crucial insights into the extent and distribution of cardiac amyloidosis.1-3 In the presented case of a 61‑year‑old man with ATTRm cardiomyopathy, scintigraphy with [99mTc]Tc‑DPD showed low myocardial uptake. The diagnostic accuracy of 99mTc‑DPD scintigraphy may be associated with the precursor protein in ATTR.4 Since ATTR amyloid fibrils contain 2 different components—full‑length TTR (type B) or a mixture of C‑terminal fragments and full‑length TTR (type A)—the affinity of 99mTc‑DPD to ATTR may be related to amyloid fibril composition and, consequently, impact scintigraphy findings.4 The presence of C‑terminal fragments, mainly seen in late‑onset ATTR V30M amyloidosis and patients with other genetic variants, is strongly linked to tracer uptake and test results.4
Beyond the TTR variant and the presence of specific types of amyloid fibrils, there are also other factors that can significantly impact the imaging findings.2 The following clinical scenarios should be considered when interpreting scintigraphy results due to their potential association with false‑positive results: apolipoprotein A‑I amyloidosis, apolipoprotein A‑II amyloidosis, apolipoprotein A‑IV amyloidosis, β2‑microglobulin amyloidosis, light‑chain amyloidosis, hydroxychloroquine cardiac toxicity, blood pool, rib fractures, valvular or annular calcification, and recent myocardial infarction (<4 weeks).2 On the other hand, false negatives may be observed in rare variants Phe84Leu and Ser97Tyr, very mild disease, and delayed or premature acquisition.2 Persistently negative scintigraphy in this case highlights the variability in sensitivity of this modality, particularly in ATTRm with low amyloid burden or specific mutations. Although false‑negative results are extremely rare, they raise concerns about the reliability of scintigraphy as a standalone diagnostic tool in this population.2 While progression of Perugini grade 1 findings to higher grades has been reported, the persistently negative results in this case suggest limitations in detecting certain hereditary subtypes.
Cardiac involvement in a majority of patients with ATTRm can be easily diagnosed with a scintigraphy‑based approach. However, it is important to consider the differences in myocardial uptake in the cases of rare pathogenic TTR gene variants. A comprehensive understanding of these intricacies is crucial for an accurate diagnosis and optimal management of patients with ATTRm cardiomyopathy.
- Maurer MS, Bokhari S, Damy T, et al. Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail. 2019;12: e006075. | Crossref
- Garcia‑Pavia P, Rapezzi C, Adler Y, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021;42: 1554‑1568. | Crossref
- Writing Committee; Kittleson MM, Ruberg FL, et al. 2023 ACC expert consensus decision pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis: a report of the American College of Cardiology Solution Set Oversight Committee. J Am Coll Cardiol. 2023; 81: 1076‑1126. | Crossref
- Pilebro B, Suhr OB, Näslund U, et al. (99m)Tc‑DPD uptake reflects amyloid fibril composition in hereditary transthyretin amyloidosis. Ups J Med Sci. 2016; 121: 17‑24. | Crossref
ARTICLE INFORMATION