“I suffer from several serious diseases.” Dunnigan type of familial partial lipodystrophy: a multiple-symptom disorder caused by a genetic mutation

 CC BY 4.0
CC BY 4.0
“I suffer from several serious diseases.” Dunnigan type of familial partial lipodystrophy: a multiple-symptom disorder caused by a genetic mutation
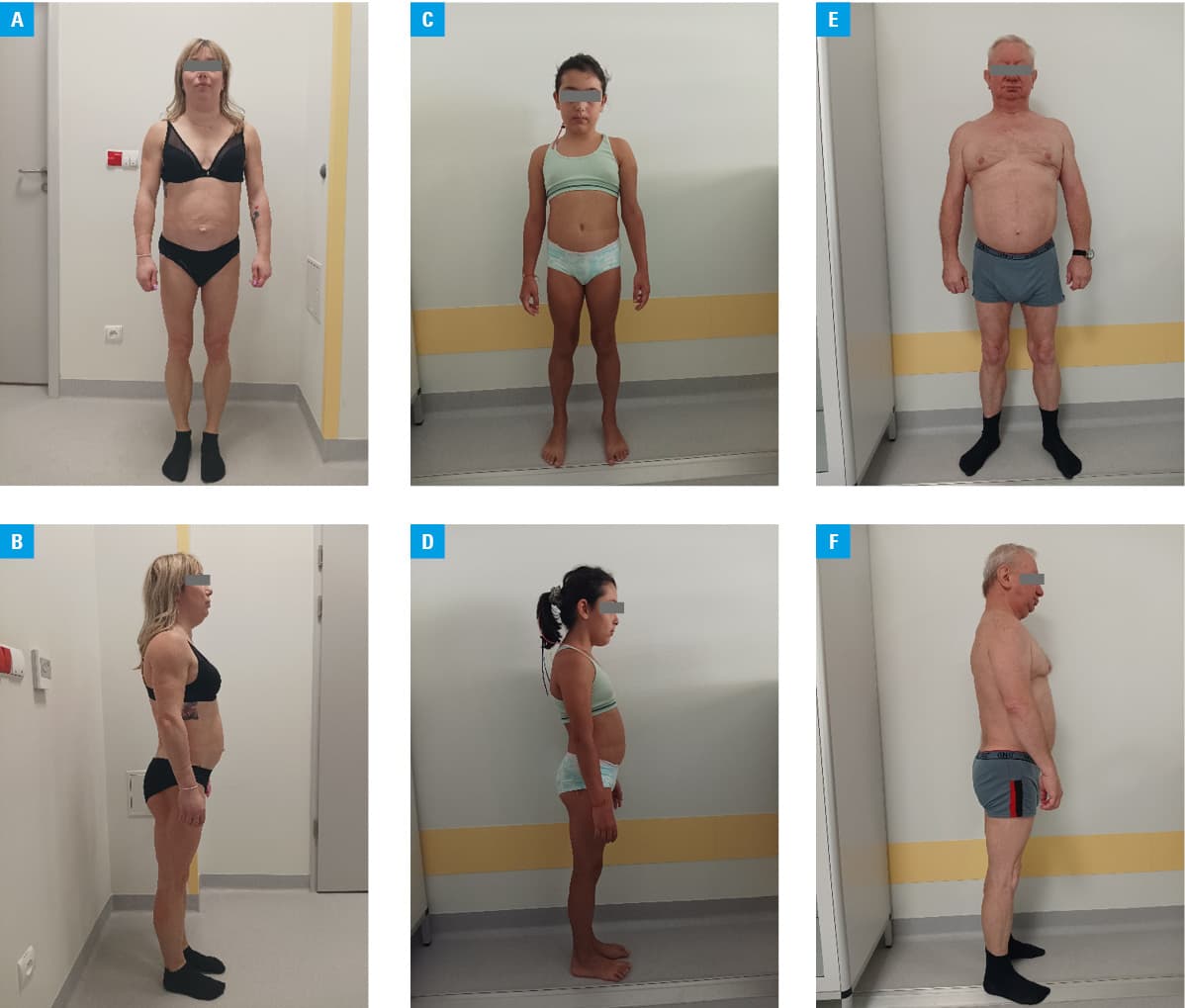
A 32‑year‑old woman was admitted to the outpatient metabolic clinic at the University Hospital in Kraków. During the visit, she referred to herself as a patient suffering from several serious diseases. Her major concern were 2 episodes of acute pancreatitis. Their most likely cause was hypertriglyceridemia (triglyceride level reached 8 mmol/l at the first episode; reference range [RR] <2.3 mmol/l).1 She was also diagnosed with type 2 diabetes. Her last therapy included oral hypoglycemic drugs (metformin and gliclazide), which did not provide satisfactory glycemic control (glycated hemoglobin, 7.2%; RR <7%). During history taking, the patient also stated that her muscular physique had attracted attention since she was a child. On physical examination, she was 156 cm tall, weighed 56 kg, and her body mass index was 23 kg/m2. Dysmorphic body structure was noticed with fat accumulation around the face, neck, and shoulders, accompanied by thin limbs with clearly visible muscles due to atrophy of the subcutaneous adipose tissue, loss of fat tissue in the region of buttocks, acanthosis nigricans in the cervical region, and hirsutism (Figure 1A and 1B). The woman informed that her 9‑year‑old daughter (Figure 1C and 1D) and father (Figure 1E and 1F) had a similar body composition. Based on the medical history that included hypertriglyceridemia, acute pancreatitis, diabetes, abnormal body composition, as well as a possible hereditary nature of the disease, familial partial lipodystrophy (FPL) was suspected.2,3 Genetic testing was performed in all 3 family members. Next‑generation sequencing genetic testing revealed a heterozygous variant in the eighth exon of the LMNA gene: c.1444C>T, p.Arg482Trp. This autosomal dominant pathogenic variant was reported in earlier publications as a cause of FPL type 2, known as Dunnigan lipodystrophy. In such cases, cascade genetic testing of the family members is advised. LMNA encodes nuclear lamins A and C, the proteins that organize nuclear architecture through structural attachments. The mutant gene products may lead to apoptosis and premature death of adipocytes.
The diagnosis of genetically confirmed Dunnigan lipodystrophy may enable consideration of new treatment options, for example, metreleptin.4 This particle is an analogue of human leptin that improves insulin sensitivity and decreases both hepatic glucose output contributing to hyperglycemia and hepatic steatosis. It is already registered in congenital generalized or acquired generalized lipodystrophy. Metreleptin is currently under investigation in a phase 3 trial for partial lipodystrophy treatment. The patient and her father are currently taking part in this clinical trial. The baseline leptin levels for these adult mutation carriers were 3.95 ng/ml and 7.48 ng/ml, respectively, while the homeostatic model assessment for insulin resistance values were 5.32 and 5.14 (RR in the homeostatic model assessment for insulin resistance <2.5), respectively, corresponding to severe insulin resistance. Triglyceride levels reached 3.57 mmol/l and 3.68 mmol/l for the patient (on fenofibrate) and her father, respectively, while it was 1.17 mmol/l in the daughter.
In summary, patients with syndromic genetic diseases with multiple clinical manifestations related to different organs and systems are frequently diagnosed many years after the initial symptoms. Careful medical interviews, including family history taking, and physical examinations are always a base for further proper diagnostics in such diseases, frequently involving genetic testing. Genetic testing in monogenic forms of metabolic diseases, such as lipodystrophy and diabetes, can currently be applied in clinical practice to establish proper diagnosis, define prognosis in family members, and, in many cases, propose tailored treatment options. Metreleptin may be registered in the future for partial lipodystrophy, improving prognosis in the affected persons.
- Lazarte J, Wang J, McIntyre AD, et al. Prevalence of severe hypertriglyceridemia and pancreatitis in familial partial lipodystrophy type 2. J Clin Lipidol. 2021; 15: 653‑657. | Crossref
- Klupa T, Szopa M, Skupien J, et al. LMNA gene mutation search in Polish patients: new features of the heterozygous Arg482Gln mutation phenotype. Endocrine. 2009; 36: 518‑523. | Crossref
- Patni N, Garg A. Lipodystrophy for the diabetologist—what to look for. Curr Diab Rep. 2022; 22: 461‑470. | Crossref
- Akinci B, Meral R, Oral EA. Update on therapeutic options in lipodystrophy. Curr Diab Rep. 2018; 18: 139. | Crossref
ARTICLE INFORMATION