Crooke cell adenoma (CCA) is a rare and aggressive subtype of Cushing disease (CD).1 Corticotroph adenomas are identified as CCAs when more than 50% of corticotroph cells present Crooke hyaline changes described as perinuclear cytokeratin deposition.1 CCAs can present clinically as functioning corticotroph adenomas, that is, CD, or clinically silent CAs.
Patients with CCA show increased mortality and difficulty with achieving long‑term remission.2 Treatment options for CCA are similar to options for standard CD. Surgery is the first‑line treatment but due to persistent postoperative hypercortisolemia, most patients would need an additional therapy (radiotherapy and medical therapy). Currently available medications are divided into pituitary‑directed therapies (somatostatin analogs, such as pasireotide or a dopamine agonist) and steroidogenesis inhibitors. Moreover, due to an aggressive nature of CCA, temozolomide was also approved for therapy. The management of CCA remains challenging.3
We present a case of a 33‑year‑old man with sudden visual impairment, initially without signs of hypercortisolemia. Pituitary magnetic resonance imaging (MRI) revealed macroadenoma (51.6 mm × 29.6 mm × 32.8 mm; Figure 1A) within the sella turcica, invading the cavernous sinus, suprasellar region, and the third ventricle, with compression of the optic chiasm. Within less than 2 months of diagnosis, sudden weight gain and striae occurred. The clinical and biochemical features of hypercortisolemia were confirmed, including high adrenocorticotropic hormone (ACTH) levels of 230 pg/ml (reference range [RR], 7.2–63.3 pg/ml), loss of circadian cortisol rhythm, elevated urinary free cortisol (UFC) excretion of 675 µg/24 h (RR, 4.3–276 µg/24 h), and a lack of cortisol suppression after administration of 1‑mg dexamethasone. Secondary hypothyroidism and hypertension were also diagnosed. The patient underwent transsphenoidal surgery, and the tumor was partially removed (around 50%; residual mass, 19 mm × 14 mm × 12 mm on MRI). However, ACTH and UFC levels did not return to normal. Histopathologic examination revealed CCA (Ki67 index <1%; p53 <1%; low O‑6‑methylguanine‑DNA methyltransferase expression in <10% cells; mitotic index, 2/10 high‑power fields).4
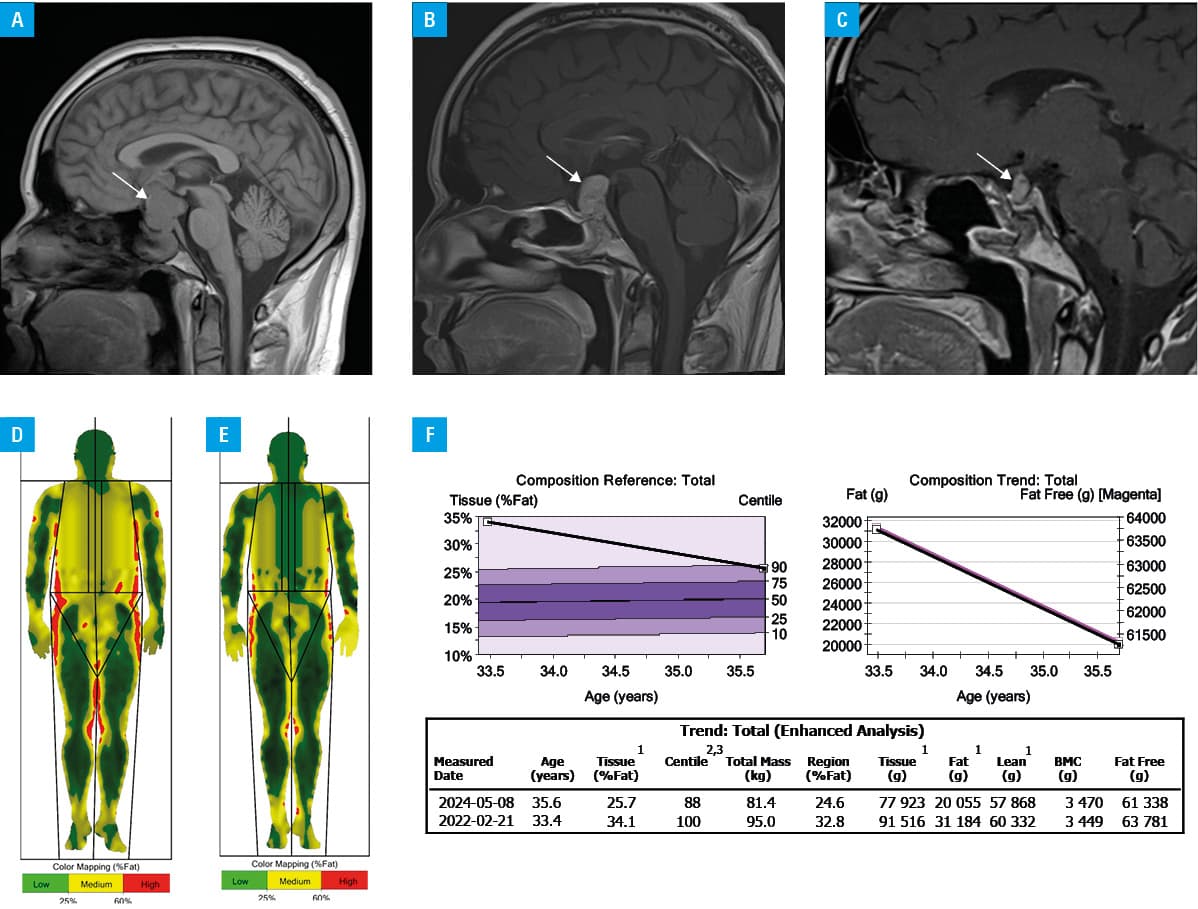
During a follow‑up hospitalization 3 months after the operation, chest computed tomography (CT) showed pulmonary embolism in the peripheral arteries, and rivaroxaban was administered. After surgery, a gradual increase in cortisol levels and tumor mass were observed. Considering the risk associated with reoperation or radiation therapy, a decision was made to introduce conservative treatment. Corticotroph adenomas express somatostatin receptors (SSTRs), mainly subtype 5 (SSTR5), followed by subtype 2 (SSTR2), and subtype 1 (SSTR1), providing potential therapeutic targets. Pasireotide is a multireceptor‑targeted second‑generation somatostatin analogue with the highest affinity for SSTR5. As the patient presented with functional CD, a long‑acting somatostatin receptor ligand pasireotide was started at an initial intramuscular dose of 10 mg, with gradual titration to 30 mg.5 Additionally, cabergoline was administered (2 mg/week). Dopamine 2 receptor (D2R) agonists are not only expressed in various pituitary cells but high D2R expression was noticed in different types of pituitary tumors, including corticotroph adenomas. However, despite 6‑month treatment, ACTH and UFC levels did not return to normal. For the next 6 months, osilodrostat (1 mg twice daily with uptitration to 4 mg per day) was added to pasireotide (30 mg) achieving the normalization of UFC. A year after treatment, MRI showed slight progression in the tumor size (22 mm × 15 mm × 20 mm; Figure 1B). Follow‑up examination confirmed diabetes, and metformin was administered. Due to a lack of improvement, the patient underwent the second transsphenoidal adenomectomy, complicated by panhypopituitarism. The UFC level normalized, but a slight increase in ACTH level persisted (150 pg/ml). Histologically, the tumor was described as densely granulated adenoma, and the Ki67 index was low. However, no Crooke hyaline change was detected. The experienced neuropathologist who performed both histopathological examinations was unable to explain these discrepancies. Typically, the tumors transform into more aggressive forms during treatment but in our case it seems to have transformed into a milder form, that is, from CCA to densely granulated corticotroph adenoma.
Pasireotide treatment was restarted, resulting in the disease stabilization (UFC within the RR of 54.6 µg/24 h and slightly elevated ACTH levels [146 pg/ml]) 1 year after the second surgery. On MRI, the tumor size was stable (residual mass, 10 mm × 7 mm × 8 mm; Figure 1C). During the 3 years of treatment, the patient’s condition improved and weight loss was noted (Figure 1D–1F). Follow‑up chest CT before the second transsphenoidal adenomectomy showed no signs of pulmonary embolism. Throughout the entire treatment with pasireotide good glycemic control was maintained with metformin.
CCA is a rare pituitary adenoma that should receive significant attention due to its aggressive nature and high recurrence rate. Data regarding the effectiveness of medical therapies in CCAs are limited. The effect of pituitary‑targeted therapy in CCAs requires further studies to clarify its efficacy in such challenging aggressive tumors, but based on the positive outcomes in our patient, pasireotide and cabergoline might be worth considering as the second‑line treatment in CCAs. Moreover, osilodrostat was shown to be effective in controlling hypercortisolism in CCA, allowing for avoidance of bilateral adrenalectomy.6 However, further studies are needed to confirm its effectiveness for this indication.
- Gilis‑Januszewska A, Wilusz M, Pantofliński J, et al. Temozolomide therapy for aggressive pituitary Crooke’s cell corticotropinoma causing Cushing’s disease ‑ a case report with literature review. Endokrynol Pol. 2018; 69: 306‑312.
- Raverot G, Burman P, McCormack A, et al. European Society of Endocrinology Clinical Practice Guidelines for the management of aggressive pituitary tumours and carcinomas. Eur J Endocrinol. 2018; 178: G1‑G24. | Crossref
- Gilis‑Januszewska A, Bogusławska A, Rzepka E, et al. Individualized medical treatment options in Cushing disease. Front Endocrinol (Lausanne). 2022; 13: 1060884. | Crossref
- Witek P, Maksymowicz M, Szamotulska K, et al. MGMT expression in pituitary corticotroph adenomas and its relationship to clinical, pathological, and ultrastructural parameters in patients with Cushing’s disease. Folia Neuropathol. 2020; 58: 357‑364. | Crossref
- Lacroix A, Gu F, Gallardo W, et al. Efficacy and safety of once‑monthly pasireotide in Cushing’s disease: a 12 month clinical trial. Lancet Diabetes Endocrinol. 2018; 6: 17‑26. | Crossref
ARTICLE INFORMATION