Expanding the phenotype of myosin heavy chain 11–related thoracic aortic disease: a case of mitral annular disjunction in a family

 CC BY 4.0
CC BY 4.0
Expanding the phenotype of myosin heavy chain 11–related thoracic aortic disease: a case of mitral annular disjunction in a family
Although the myosin heavy chain 11 (MYH11) gene is definitively linked to thoracic aortic aneurysms and dissections (TAADs), the frequency of pathogenic / likely pathogenic MYH11 variants among patients with TAAD is below 1%. Therefore, our knowledge of this rare disease is scant. MYH11-related disease is associated with TAAD, patent ductus arteriosus (PDA), and cerebral arteriopathies.1-4
We report a case of a 2‑generation family (4 affected individuals) with TAAD associated with the MYH11 p.Arg712Gln variant. A 37‑year‑old woman, with a family history of TAAD, reporting heart palpitations and atypical chest pain, was referred to our unit. Her blood pressure was normal. Transthoracic echocardiography showed mild dilatation of the aortic root and ascending aorta. Additionally, moderate mitral regurgitation due to bileaflet mitral valve prolapse (MVP) and mitral annular disjunction (MAD) with a disjunction length of 8 mm (Figure 1A–1D) was observed. Computed tomography angiography of the entire aorta confirmed the “extended phenotype” of aortic dilatation, with an aortic root diameter of 38 mm and maximal thoracic aorta diameter of 40 mm. Furthermore, she had ventricular arrhythmia, with 1193 premature ventricular complexes per day.
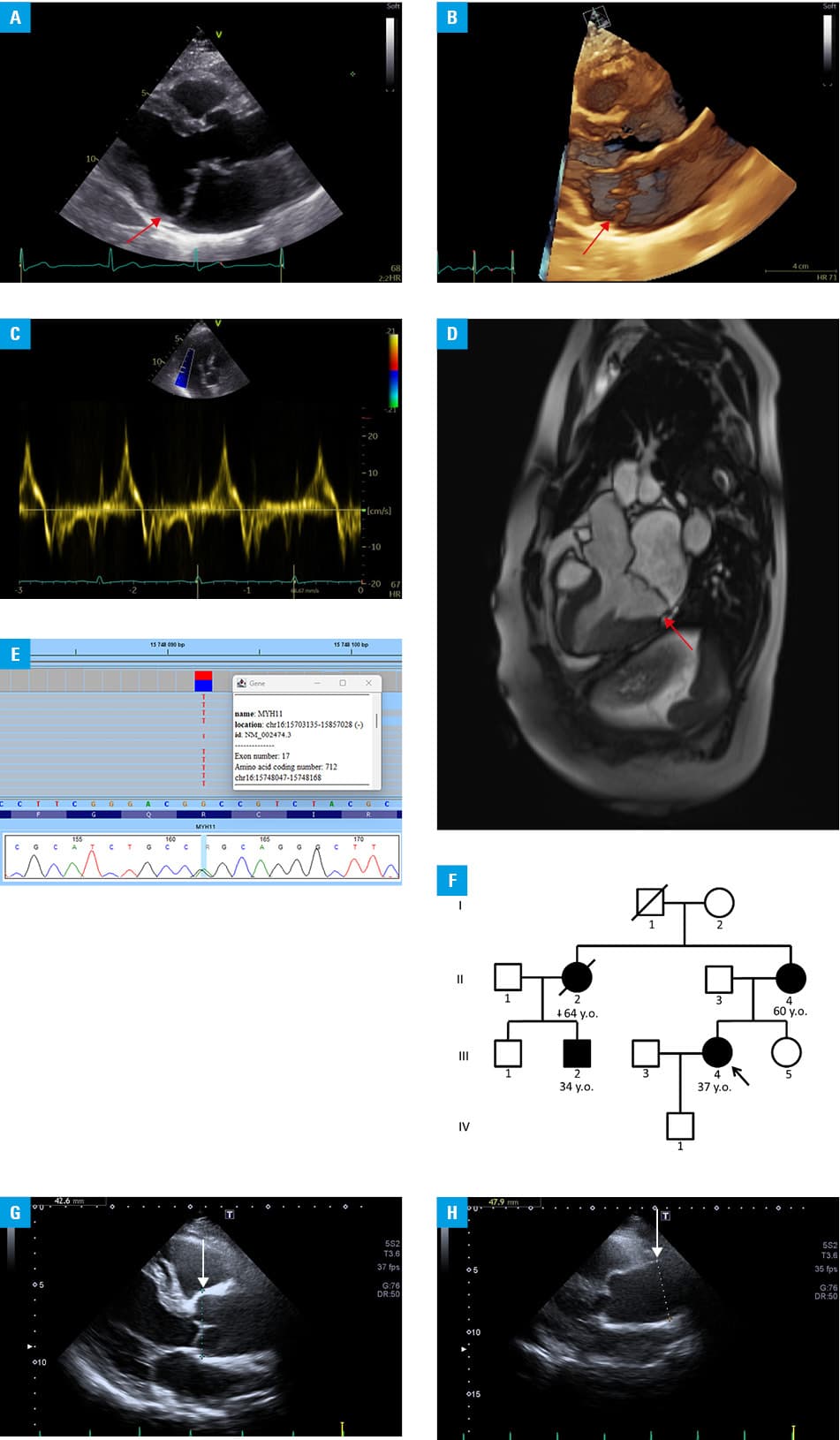
Abbreviations: y.o., years old
The patient had a positive family history of the David procedure performed on her mother at the age of 60 years due to thoracic aortic aneurysm (aortic root of 49 mm and ascending aorta diameter of 56 mm) along with significant aortic valve insufficiency. Her mother’s sister developed acute thoracic aortic dissection type A at the age of 62 years and died of cancer 2 years later. These 3 cases in a single family prompted a search for the genetic cause of TAAD. Genetic testing identified an NM_002474.3:c.2135G>A transition in the MYH11 gene (Figure 1E), resulting in a missense substitution (p.Arg712Gln) in the ATPase head region. This variant is classified as pathogenic according to the American College of Medical Genetics criteria.5 Following identification of the pathogenic MYH11 variant, cascade phenotypic and genetic screening was performed in the proband’s relatives (Figure 1F). The same MYH11 variant was found in the patient’s affected mother and her affected sister’s son, confirming that the affected maternal sister was an obligate carrier. The proband’s cousin (Figure 1G and 1H) exhibited a similar extended phenotype of ascending thoracic aortic dilatation, with an aortic root diameter of 42 mm and a tubular segment of 47 mm. Unlike in the patient’s family, the p.Arg712Gln variant in a study by Pannu et al3 was associated with TAAD, PDA, and premature occlusive vascular disease.
It remains unclear whether mitral and aortic diseases are causally linked, as MAD was found only in a single affected carrier. Although the prevalence of MVP in the general population is estimated at 2%–3%, the prevalence of MAD is unknown. In the Montalcino Aortic Consortium study,6 the prevalence of MVP among patients with heritable TAAD was much higher, namely, 15%, and 1 in 10 patients with MVP had MAD. Of note, MAD was predominantly associated with syndromic TAAD, such as Marfan and Loeys‑Dietz syndromes, and was described in only 1 case with the MYH11 variant.6 To our knowledge, this is the first report of familial TAAD due to the MYH11 variant with coexisting MAD. The extended phenotype of aortic dilatation was consistently present in all carriers, and thoracic aortic events were observed beginning in the seventh decade of life.
- Poninska JK, Bilinska ZT, Franaszczyk M, et al. Next‑generation sequencing for diagnosis of thoracic aortic aneurysms and dissections: diagnostic yield, novel mutations and genotype phenotype correlations. J Transl Med. 2016; 14: 1‑17. | Crossref
- Zhu LM, Vranckx R, Khau Van Kien P, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm / aortic dissection and patent ductus arteriosus. Nat Genet. 2006; 38: 343‑349. | Crossref
- Pannu H, Tran‑Fadulu V, Papke CL, et al. MYH11 mutations result in a distinct vascular pathology driven by insulin‑like growth factor 1 and angiotensin II. Hum Mol Genet, 2007. 16: 2453‑2462. | Crossref
- Strzelczyk J, Sobieraj P, Placha G, Jędrusik P. Multiple intracranial aneurysms and fibromuscular dysplasia of renal arteries in a woman with a variant of the myosin heavy chain 11 gene. Pol Arch Intern Med. 2023; 133: 16424. | Crossref
- Richards S, Aziz N, Bale S, et al; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17: 405‑424. | Crossref
ARTICLE INFORMATION