B-cell lymphoma or Crohn disease? Navigating diagnostic ambiguity in small intestine pathology

 CC BY 4.0
CC BY 4.0
B-cell lymphoma or Crohn disease? Navigating diagnostic ambiguity in small intestine pathology
A 49‑year‑old man presented with a 10‑year history of recurrent epigastric pain and persistent mild iron deficiency anemia over the past 3 years. Initial symptoms included mild dyspepsia and peripheral arthralgia, with no significant gastrointestinal bleeding or changes in bowel habits. Proton pump inhibitors initially provided symptomatic relief.
Between 2014 and 2023, multiple diagnostic procedures were performed, including 8 upper gastrointestinal endoscopies and 2 ileocolonoscopies. Recurrent findings included duodenal ulcers without Helicobacter pylori infection, while lower gastrointestinal tract examinations showed minor abnormalities (small, polypoid elevations of the mucosa in the terminal ileum). Histological examination of duodenal and ileal biopsies indicated nonspecific chronic lymphoplasmacytic infiltrates and mild villous architectural changes, raising concerns for potential Crohn disease and celiac disease. Biopsy of a millimeter‑sized cecal polyp demonstrated monomorphic B‑cell infiltrates with unusual immunohistochemical profile and high Ki‑67 proliferation.
Laboratory tests highlighted multiple abnormalities suggestive of a nonspecific autoimmune tendency, including seropositivity for anti–Saccharomyces cerevisiae antibodies and perinuclear antineutrophil cytoplasmic antibodies without clinical symptoms directly correlating with the immunologic profile. Notably, fecal calprotectin levels increased from 250 µg/g to 1000 µg/g (reference range <50 µg/g), despite a lack of macroscopic signs of colonic inflammation. Genetic and serologic tests for celiac disease were negative.
Computed tomography (CT) and magnetic resonance enterography demonstrated mesenteric lymphadenopathy and borderline splenomegaly without visible intestinal lesions. Capsule endoscopy identified polypoid lesions in the proximal jejunum and a solitary erosion in the terminal ileum. Bone marrow biopsy and positron emission tomography/CT showed no signs of pathology or systemic disease. Finally, enteroscopy was performed and 2 ulcers in the proximal ileum (one oval, 6 mm in diameter and one oblong, transversely situated, 15 mm long × 6 mm wide, surrounded by hyperemic mucosa were detected (Figure 1A).
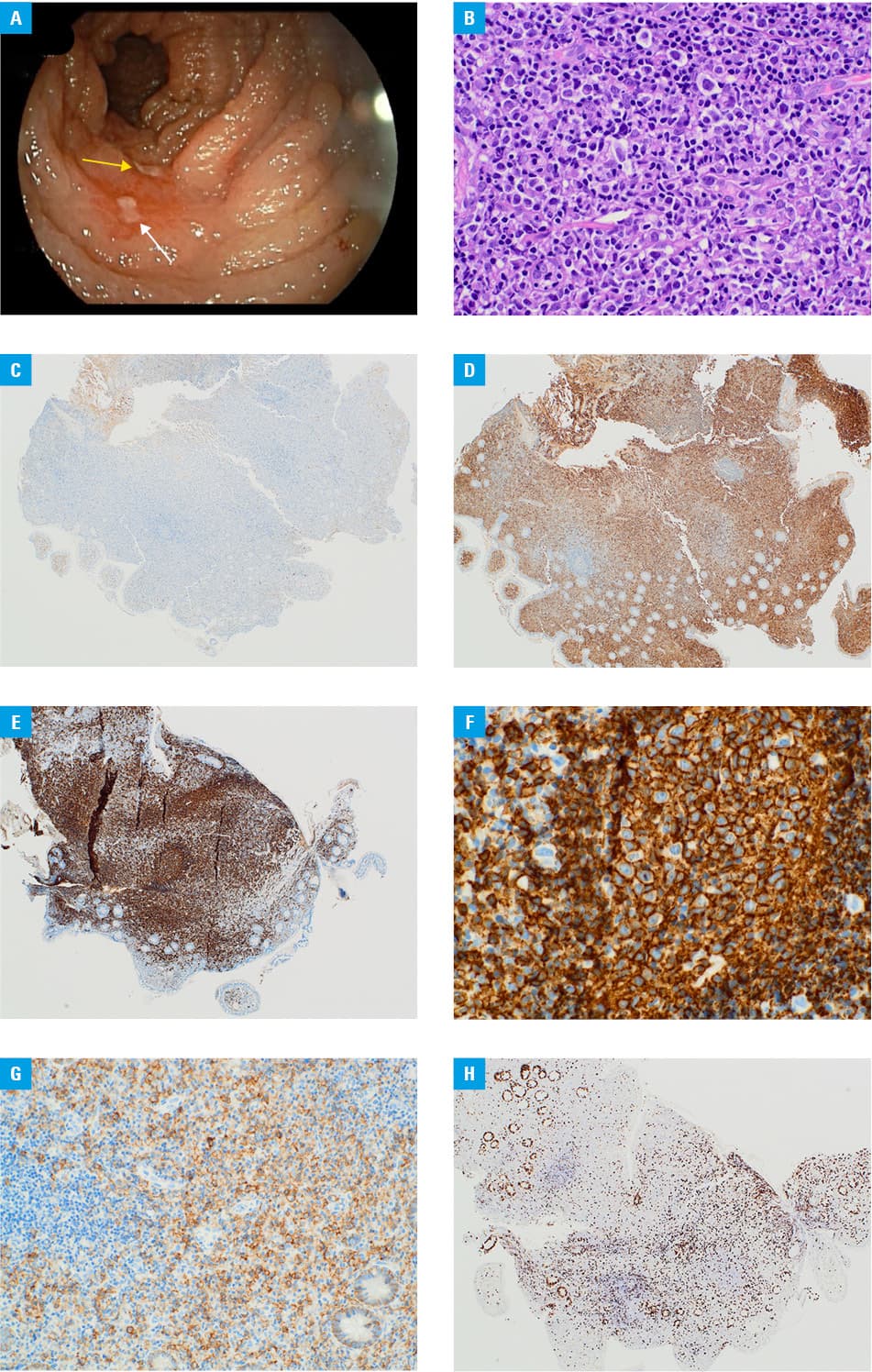
The endoscopic macroscopic examination suggested Crohn disease, while the histopathologic report provided a brief description of chronic ileitis. Given the inconclusive nature of microscopic findings and persistently abnormal biopsy material, along with a lack of definitive diagnosis and ongoing anemia, histopathologic consultation was ordered.
The examination found lymphoplasmacytic infiltration, replacing the glandular tissue with reactive germinal centers and reduced crypt architecture (Figure 1B). Immunohistochemical analysis confirmed sparse B‑cells (CD20+/PAX5+/CD23–/cyclinD1–) and plasma cells (CD138+/CD20–/PAX5–/CD56–/cyclinD1–/κ–/λ+) with low proliferative activity (Ki‑67 <10%) (Figure 1C–1H).
A secondary pathology review at the Icahn School of Medicine (New York, New York, United States) ruled out lymphoplasmacytic lymphoma based on the absence of the MYD88 L265P mutation and limited B‑cell infiltration. Therefore, 2 independent pathologists concurred, diagnosing the patient with extranodal marginal zone lymphoma (EMZL).
The patient was referred to a hematologist for further treatment. Considering the indolent yet progressive nature of the disease, with the clinical classification of stage IV lymphoma, a conservative therapeutic approach with chemotherapy was selected.
This case underscores the critical role of multidisciplinary collaboration in establishing a definitive diagnosis. For suspected cases of Crohn disease and seronegative celiac disease, regular assessment of therapeutic response is crucial, particularly in the cases of atypical presentation or location.1-5 Key diagnostic insights emerged through repeated endoscopy, in particular biopsy, and immunohistochemistry, ultimately identifying EMZL as a culprit. Without proactive and iterative testing, the diagnosis might have remained elusive.
- Di Rocco A, Petrucci L, Assanto GM, et al. Extranodalmarginal zone lymphoma: pathogenesis, diagnosis and treatment. Cancers (Basel). 2022; 14: 1742. | Crossref
- Burke JS. Lymphoproliferative disorders of the gastrointestinal tract: a review and pragmatic guide to diagnosis. Arch Pathol Lab Med. 2011; 135: 1283‑1297. | Crossref
- Badwaik N, Gharde P, Lamture Y, et al. A rare case of mucosa‑associated lymphoid tissue lymphoma in the ileum. Cureus. 2022; 14: e32851. | Crossref
- Da B, Zhang J, Zhu F, et al. Extranodal marginal zone lymphoma of mucosa‑associated lymphoid tissue of the ileum in an adult presenting with intussusception: a case report and literature review. Front Oncol. 2024; 14: 1395144. | Crossref
- Markopoulos K, Bührer E, Banz Y, et al. Challenges in the diagnosis of marginal zone lymphoma with symptoms of small intestinal disease: a case report and scoping review of the literature. J Gastrointest Oncol. 2022; 13: 2583‑2607. | Crossref
ARTICLE INFORMATION