New therapies in multiple myeloma: benefits and limitations
Key words: bispecific antigen, chimeric antigen receptor T cell, cytokine release syndrome, immune effector cell–associated neurotoxicity syndrome, multiple myeloma



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
New therapies in multiple myeloma: benefits and limitations
Multiple myeloma (MM) is a bone marrow cancer caused by clonal proliferation of plasma cells, and it is classified as a plasma cell dyscrasia. Over the last 20 years, there has been a dramatic increase in the availability of new therapies for patients with MM with a significant improvement in remission duration and overall survival.
Introduction: of immunomodulatory drugs (thalidomide, lenalidomide, pomalidomide), monoclonal antibodies (eg, daratumumab), and proteasome inhibitors (bortezomib, karflizomib) into the treatment paradigm has meaningfully and favorably changed the prognosis for MM patients. More recently, development of molecular‑targeted therapies, especially bispecific antibodies and chimeric antigen receptor (CAR)-T cells as part of immunotherapy has rapidly evolved into their use in clinical practice. This review provides an overview of emerging molecular‑targeted therapies for MM, highlighting bispecific antibodies and CAR T‑cell approaches, and examining key structural and functional considerations, principal findings from current clinical trials, and strategies for managing therapy‑related toxicities.
Introduction
Multiple myeloma (MM) is caused by clonal proliferation of plasma cells (PCs), typically with an associated monoclonal gammopathy, and idiotypic rearrangement of the immunoglobulin (Ig) gene preceding malignant transformation of a plasma precursor cell. Table 1 presents the current International Myeloma Working Group criteria for the diagnosis of MM. When an emerging cell clone reaches a size of at least 5 × 10⁹ cells, its product, monoclonal Ig, also known as monoclonal protein (M protein), becomes visible as an abnormal peak in a serum protein electrophoresis gel. MM accounts for approximately 1.8% of adult cancer diagnoses and 10% of hematologic malignancy diagnoses.1-5 The median age at diagnosis is approximately 65–70 years.6-8 According to the 2016–2020 Surveillance, Epidemiology, and End Results data, the percentages of new cases by age group are: 0.4% for 20–34 year olds, 2.7% for 35–44 year olds, 9.7% for 45–54 year olds, 22.7% for 55–64 year olds, 31.9% for 65–74 year olds, 23.8% for 75–84 year olds, and 8.8% for over 84 year olds. Median age of death is approximately 75 years and overall survival (OS) at 5 years is 61%. In 2020, 176 404 (95% uncertainty interval [UI], 167 933–185 303) new cases of MM were reported worldwide.9 In the United States (USA), more than 35 000 new cases of MM are diagnosed each year, and approximately 13 000 patients die from the disease.10 From 2000 to 2020, 217 049 cases of MM were reported in the USA with the majority being men (54.85%) over 55 years of age, and African Americans being affected twice as often as white people.9,11 Additionally, the age‑standardized incidence rate was 1.92 per 100 000 persons (95% UI, 1.68–2.12) in 2019, as compared with 1.72 per 100 000 persons (95% UI, 1.59–1.93) in 1990, indicating a significant increase in incidence over the last 30 years.7 Globally, the incidence of MM varies by region, with the highest rates observed in Australia, New Zealand, North America, and Western Europe, and the lowest in Asia.12 Based on data from the National Cancer Registry in Poland, the number of new cases of MM in 2019 was 1713, affecting 808 men and 905 women, with MM being responsible for 683 deaths among men and 727 among women.
Abbreviations: BM, bone marrow; CT, computed tomography; LLN, lower limit of normal; MRI, magnetic resonance imaging; PET, positron emission tomography; ULN, upper limit of normal | ||
Requirement | Criteria | Description |
Clonal plasma cells or plasmacytoma | Mandatory |
|
Myeloma‑defining events | At least 1 of the following:
| |
CRAB description | ||
C – hypercalcemia | Serum calcium >11 mg/dl (2.75 mmol/l) OR
>1 mg/dl above ULN | |
R – renal insufficiency | Serum creatinine >2 mg/dl (177 µmol/l) OR
creatinine clearance <40 ml/min | |
A – anemia | Hemoglobin <10 g/dl OR
>2 g/dl below LLN | |
B – bone lesions | One or more osteolytic lesions on skeletal radiography, CT, or PET‑CT | |
Biomarkers of malignancy | ||
Clonal bone marrow plasma cells | ≥60%, as detected on bone marrow biopsy | |
Involved / uninvolved free light chain ratio | Ratio ≥100 and involved light chain ≥100 mg/l | |
Focal lesions on MRI | >1 focal lesion >5 mm in size | |
Development of multiple myeloma
Patients diagnosed with MM evolve from the premalignant stage termed monoclonal gammopathy of undetermined significance (MGUS). MGUS is asymptomatic and is present in approximately 3%–5% of the population over the age of 50 years. It may evolve into MM, light‑chain amyloidosis, or Waldenström macroglobulinemia.13-15 On average, approximately 1% of people with MGUS progress to MM or a related disorder each year16 according to models based upon the level of M protein, Ig isotype, and free light‑chain ratios that predict the likelihood of evolving into MM. MGUS is 3 times more common in African American patients than in white patients.17 Smoldering multiple myeloma (SMM) is an asymptomatic clonal PC disorder that represents an intermediate stage between MGUS and MM. No single pathological, molecular, or clinical feature can reliably distinguish these 2 groups of patients.18 However, in everyday clinical practice, distinguishing SMM from MGUS is important, as the risk of progression of SMM to MM is 10 times higher than that of MGUS to MM in the first 5 years following diagnosis (10% per year in SMM vs 1% per year in MGUS).19
Current standards of care
Modern therapeutic landscape for MM is shaped by our increasingly nuanced understanding of the disease’s underlying biology and emergence of a diverse array of targeted and immunologically driven therapies. Historically, patients received treatments such as melphalan and prednisone, dexamethasone, and / or high‑dose chemotherapy followed by autologous stem cell transplantation (ASCT), but relapses were inevitable, and survival proved very limited. The advent of novel agents—specifically immunomodulatory drugs (IMiDs), proteasome inhibitors (PIs), and monoclonal antibodies (mAbs)—substantially changed clinical outcomes and laid the groundwork for the next generation of MM management treatment strategies. Today, quadruplet regimens that integrate a CD38‑directed mAb, a PI, an IMiD, and dexamethasone have become a mainstay for many patients in the upfront setting, and in particular those who are eligible for transplant as well as the transplant ineligible population. At the same time, further advances including next‑generation small molecules, peptide‑drug conjugates, antibody‑drug conjugates (ADCs), and most recently cereblon E3 ligase modulators (CELMoDs) promise further gains, especially in the relapsed / refractory setting where therapeutic options must be both effective and noncross resistant, with manageable toxicity.
Evolution of frontline therapy and the role of quadruplet regimens
A critical turning point in frontline MM therapy was a finding that combining an IMiD (such as lenalidomide) with a PI (eg, bortezomib) and dexamethasone significantly improved remission rates, leading to higher percentages of patients achieving deep responses and improved outcomes. These 3‑drug (triplet) regimens replaced older doublets, becoming a global standard of care for newly‑diagnosed MM (NDMM) both in transplant eligible and transplant ineligible populations.20 This approach was further enhanced upon introducing CD38‑targeting mAbs, with daratumumab at the forefront and now isatuximab. CD38 is a glycoprotein highly and uniformly expressed on PCs, making it an attractive target for selective immunotherapy. Early clinical studies of daratumumab in heavily pretreated populations revealed its single‑agent potency, prompting subsequent trials that placed it alongside bortezomib, lenalidomide, and dexamethasone (RVd) and also carfilzomib‑based (carfilzomib, lenalidomide, dexamethasone [KRd]) therapy. The Dara‑RVd quadruplet regimen has demonstrated superior depth of response, longer progression‑free survival (PFS), and higher minimal residual disease (MRD) negativity rates than the triplet backbone of RVd alone.21 Data from trials such as GRIFFIN (Study Comparing Daratumumab, Lenalidomide, Bortezomib, and Dexamethasone [D‑RVd] Versus Lenalidomide, Bortezomib, and Dexamethasone [RVd] in Subjects With Newly Diagnosed Multiple Myeloma) and PERSEUS (Daratumumab, Velcalde [Bortezomib], Lenalidomide and Dexamethasone Compared to Velcalde, Lenalidomide and Dexamethasone in Subjects with Previously Untreated Multiple Myeloma) established this approach, showing that daratumumab‑based quadruplet therapies not only induce higher rates of stringent complete response (sCR) but also correlate with meaningful improvements in PFS.21,22 In patients with high‑risk cytogenetics, including those harboring del(17p) or t(4;14) mutations, these more intensive inductions have generated deeper responses and helped mitigate the poor prognostic impact traditionally associated with these cytogenetic abnormalities.22 For transplant‑eligible patients, the integration of quadruplet induction followed by high‑dose therapy and ASCT remains a key strategy, with most institutions later implementing maintenance therapy (typically with lenalidomide, sometimes in combination with a mAb and / or PI) to prolong remission. Quadruplet therapy, therefore, has become emblematic of the modern frontline approach, aiming to deliver powerful cytoreduction that sets the stage for durable disease control.
Additional monoclonal antibodies in the CD38 and SLAMF7 space
While daratumumab established the importance of targeting CD38 in MM, isatuximab further validated this target. Isatuximab, similarly to daratumumab, relies on antibody‑dependent cellular cytotoxicity, complement‑dependent cytotoxicity, and direct proapoptotic signals. In combination with agents such as carfilzomib, lenalidomide, and dexamethasone in NDMM patients and especially in high‑risk individuals, this quadruplet has proved highly effective. According to previous studies comparing the use of isatuximab with carfilzomib and dexamethasone (Isa‑KD) or isatuximab with pomalidomide and dexamethasone (Isa‑PD) in the relapsed setting, isatuximab has repeatedly shown activity in patients with advanced disease.23-25 Its different epitope binding and pharmacologic profile in comparison with daratumumab offer clinicians additional therapeutic flexibility, especially in patients who have already received daratumumab‑containing regimens or with specific disease characteristics, such as 1q amplification. Moreover, the availability of subcutaneous administration of these antibodies has significantly reduced infusion‑related reactions and decreased chair times, further improving patient quality of life.26 Elotuzumab, a monoclonal antibody that targets SLAMF7, has a distinctly different mechanism of action. SLAMF7 is expressed by malignant PCs and natural killer (NK) cells but is generally absent on other normal cell types. This enables elotuzumab to activate NK‑cell function, while disrupting myeloma cell adhesion within the bone marrow microenvironment.27 Combinations of elotuzumab with lenalidomide or pomalidomide and dexamethasone have shown durable responses in the relapsed setting. Though elotuzumab may be less commonly used in frontline settings than either daratumumab‑based or isatuximab‑based quadruplets, it remains a valuable option in certain relapsed / refractory cases, especially where immunomodulatory mechanisms are pivotal for disease control.28,29
Autologous stem cell transplantation, the role of maintenance, and future directions in an evolving landscape
Despite the influx of novel agents, ASCT retains a central role in MM therapy for transplant‑eligible patients. Studies consistently show that high‑dose melphalan followed by ASCT deepens response rates, often driving patients into MRD negativity—a key predictor of prolonged PFS and possibly OS, although the lack of OS benefit in recent studies of early ASCT, as compared with novel therapy, points to both competing risk and increasing efficacy of salvage strategies. Maintenance strategies post‑ASCT commonly include lenalidomide, although the combination of lenalidomide with a CD38‑targeting antibody or a second‑agent PI is increasingly tested, aiming to extend remission durability.30,31 As quadruplet induction regimens, innovative immunotherapies, and new targeted agents continue to advance, further questions arise about the optimal timing of ASCT, with multiple additional trials now investigating delayed transplantation in patients who achieve deep remissions via novel therapies alone. Emerging research is also evaluating whether post‑transplant therapy may incorporate T‑cell redirections or ADC in MRD settings, potentially eradicating clonal disease at a point of low tumor burden. Personalized medicine approaches, leveraging genomic and immunologic profiling, are likely to guide these decisions. Biomarkers, including cytogenetics, gene expression signatures, and MRD status assessed by next‑generation sequencing or flow cytometry, are poised to inform which patients might benefit from intensifying or deintensifying certain therapies. The phase 3 ATLAS trial (Trial of Carfilzomib, Lenalidomide, Dexamethasone Versus Lenalidomide Alone After Stem‑cell Transplant for Multiple Myeloma) evaluated maintenance therapy with KRd vs lenalidomide alone in patients with NDMM post–autologous hematopoietic stem cells transplantation (HSCT). At median follow‑up of 33.8 months, median PFS was 59.1 months with KRd vs 41.4 months with lenalidomide alone (hazard ratio, 0.51; P = 0.012). MRD negativity after 6 cycles was achieved in 44% of patients receiving KRd vs 27% with lenalidomide alone (P = 0.027). These findings suggest that KRd maintenance may offer superior PFS and higher MRD negativity rates in this patient population.32
Bispecific T‑cell engagers: teclistamab, elranatamab, and beyond
In parallel with CD38 and SLAMF7 mAbs, a major leap in immunotherapy came with bispecific T‑cell engagers (TCEs). These agents are engineered to bind simultaneously to a tumor‑associated antigen on myeloma cells (often B‑cell maturation antigen [BCMA]) and CD3 on T cells, catalyzing a targeted immune synapse that triggers T‑cell mediated cytotoxicity. Unlike CAR‑T cells—which require complex ex vivo manipulation, patient‑specific manufacturing, and extended lead times—TCEs are “off‑the‑shelf” therapies. This means they can be administered rapidly and repeated as necessary. Teclistamab emerged as a leading BCMA‑directed TCE, boasting high response rates among patients who have exhausted standard classes of therapy, including PIs, IMiDs, and CD38 mAbs. While cytokine release syndrome (CRS) and immune effector cell–associated neurotoxicity syndrome (ICANS) can occur, step‑up dosing schedules and vigilant monitoring have significantly improved the tolerability profile.33,34 Other BCMA‑directed TCEs, such as elranatamab, are advancing through clinical trials with similarly high overall response rates (ORR). Non‑BCMA targets have also entered the arena, most notably talquetamab (G protein–coupled receptor class C group 5 member D [GPRC5D] × CD3), which has showed strong activity in patients who have progressed on BCMA‑based strategies.34 Although talquetamab has a unique adverse event profile—including cutaneous, nail, and taste disturbances—ongoing dose optimization, prophylaxis, and toxicity management strategies aim to refine its therapeutic index.35 In Poland, the following bispecific antibody (BsAb) therapies are currently available through the national drug program: teclistamab, talquetamab, and elranatamab. These agents are indicated in patients with relapsed / refractory MM (RRMM) who have received at least 3 prior lines of therapy (LOT), including an IMiD, PI, and an anti‑CD38 mAb.
Antibody‑drug conjugates: belantamab mafodotin, and other next‑generation agents
ADCs combine the specificity of a mAb with the potent cytotoxicity of a chemotherapy payload. This combination allows for targeted killing of malignant cells while reducing collateral damage to healthy tissues. Belantamab mafodotin, targeting BCMA, was the first ADC of its kind to demonstrate robust activity in patients with triple‑class refractory MM.36 Encouraging results from the DREAMM‑2 study (A Study to Investigate the Efficacy and Safety of Two Doses of GSK2857916 in Participants with Multiple Myeloma Who Have Failed Prior Treatment With an Anti‑CD38 Antibody), including durable responses, brought the drug to market in certain regions; however, ocular toxicities, particularly keratopathy, posed significant challenges to long‑term use.37,38 Even so, belantamab mafodotin remains under active investigation in combination regimens (such as with PIs or IMiDs) and is provided at modified doses to balance efficacy against corneal side effects, with strict clinical benefit seen in continuation with bortezomib in the DREAMM‑7 trial (Japan Expansion Cohort: Evaluation of Efficacy and Safety of Belantamab Mafodotin, Bortezomib and Dexamethasone Versus Daratumumab, Bortezomib and Dexamethasone in Participants with Relapsed / Refractory Multiple Myeloma) and a similarly impressive clinical benefit in the DREAMM‑8 study (Belantamab Mafodotin Plus Pomalidomide and Dexamethasone (Pd) Versus Bortezomib Plus Pd in Relapsed / Refractory Multiple Myeloma) with pomalidomide.39,40 Apart from belantamab, other ADCs are being developed to target GPRC5D, reflecting ongoing efforts to expand this therapeutic class. These next‑generation ADCs employ novel linkers, payloads, or antibodies, aiming to achieve higher response rates, maintain durability, and minimize off‑target toxicities. As combination therapies become more prevalent, ADCs may be used synergistically with IMiDs or CELMoDs to target the disease from multiple angles. In a similar context, a peptide drug conjugate melfufen provides a novel aminopeptidase targeted approach to directly deliver a cytotoxic warhead to a tumor, thus sparing normal tissue. It has been approved for the treatment of relapsed / refractory disease in Europe based upon positive results from the HORIZON trial (A Study of Melphalan Flufenamide [Melflufen] Plus Dexamethasone in Patients with Relapsed or Refractory Multiple Myeloma) and the OCEAN study (A Study of Melphalan Flufenamide [Melflufen]-Dex or Pomalidomide‑dex for RRMM Patients Refractory to Lenalidomide). Melflufen has also showed promise in combination with both bortezomib and daratumumab.41-44
Cereblon E3 ligase modulators: extending the concept of immunomodulatory drugs
IMiDs revolutionized MM treatment by binding to cereblon, altering the function of an E3 ubiquitin ligase complex, and leading to selective degradation of transcription factors (Ikaros and Aiolos) integral to myeloma cell growth.45 CELMoDs, exemplified by iberdomide (CC‑220) and mezigdomide (CC‑92480), are engineered to enhance these effects. They display greater affinity for cereblon, promoting more pronounced degradation of target proteins, which can translate into both direct cytotoxic effects against myeloma cells and enhanced immune stimulation via T and NK cells. Early‑phase clinical trials have yielded very promising efficacy data, even in heavily pretreated populations resistant to prior IMiDs, PI, and BCMA‑based therapies. Mezigdomide has showed an encouraging ORR, including deep responses such as CR, with a manageable toxicity profile dominated by hematologic adverse events, namely neutropenia and thrombocytopenia.46 Iberdomide demonstrates a similar therapeutic index, and has sparked interest as a combination partner with established regimens.45 Taken together, these findings signal that CELMoDs could become integral components of future MM treatment, moving beyond salvage therapy into earlier LOT once their efficacy and safety in combination approaches are more clearly defined in the context of ongoing phase 3 clinical trials.47
Cell therapies: general information
The improvement in treatment and outcomes in MM over the past 20 years is unprecedented. Life expectancy has improved significantly, and MM is now considered a chronic disease in many patients. Before the introduction of CAR‑T–cell therapies and TCEs, patients with myeloma refractory to anti‑CD38 mAbs, IMiDs, and PIs (triple‑class refractory patients) had survival rates of less than 1 year, and patients with penta‑class refractory (anti‑CD38 mAbs, 2 IMiDs, and 2 PIs) disease usually survived less than 6 months.48 Due to the ever‑growing population of triple‑class and penta‑class refractory patients, there is still an unmet need for new treatment options to improve OS, which has contributed to the development of new therapeutic modalities.49 Treatment history of MM and current therapeutic landscape are summarized in Figure 1.
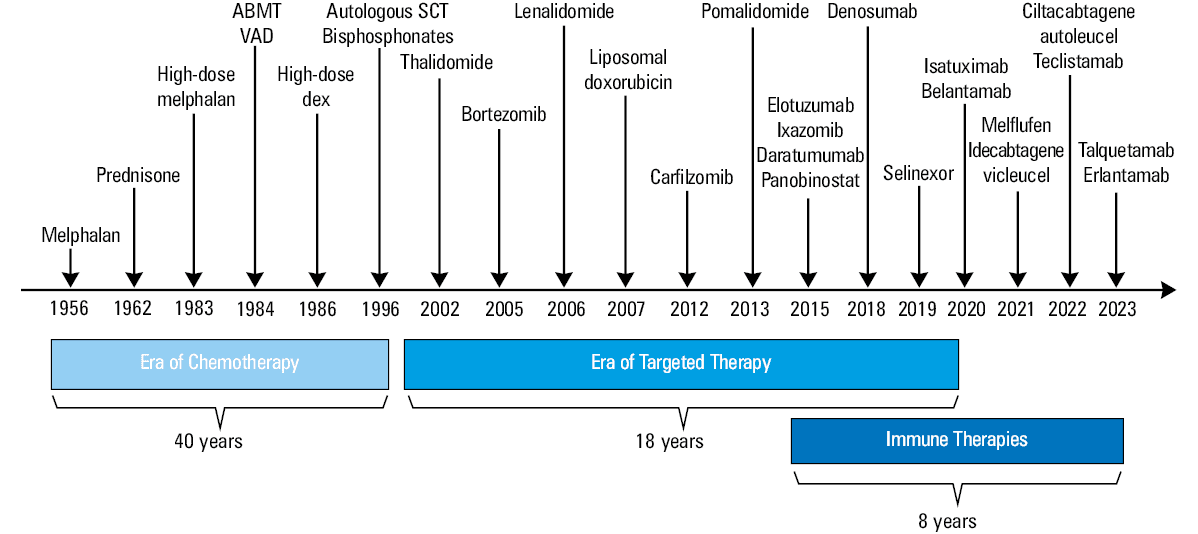
Abbreviations: ABMT, autologous bone marrow transplantation; dex, dexamethasone; SCT, stem cell transplantation; VAD regimen, vincristine (V), doxorubicin (A), dexamethasone (D)
New antigens as targets
B‑cell maturation antigen
BCMA (also known as CD269, or tumor necrosis factor receptor superfamily member 17) is expressed at high levels in both normal and malignant PCs and is a target for novel MM therapies. According to the latest gene and protein expression profiles, BCMA is the most selectively expressed cell surface receptor on MM cell lines and MM patient cells.50 BCMA is expressed in MM cells, plasma blasts, mature PCs, and late memory B cells, but it is not expressed in hematopoietic stem cells or nonhematopoietic cells.51 In everyday clinical practice, an important aspect is the loss of BCMA expression, which can lead to resistance to the applied treatment. The membrane‑bound BCMA receptor is removed from the PC by gamma secretase, leading to an increase in circulating soluble BCMA. This results in a decrease in the binding of a proliferation‑inducing ligand (APRIL) and B‑cell–activating factor (BAFF) to BCMA and a decrease in target availability for targeted therapy.52 BAFF (also called BLyS or TALL‑1) and APRIL are cytokines belonging to the tumor necrosis factor (TNF) ligand superfamily. They play important roles in B‑cell maturation, selection, and survival.53 BAFF and APRIL bind with different affinity to 3 receptors found on B lymphocytes: BCMA, transmembrane activator and cyclophilin ligand interactor (TACI), and BAFF receptor (BAFF‑R). BAFF‑R is a ligand for BAFF, while TACI and BCMA bind to both APRIL and BAFF. Therefore, BAFF and APRIL require an appropriate amount of BCMA for their proper function. The role of BAFF‑R includes participation in the formation of germinal centers, as well as the selection and survival of B cells. Additionally, similar to TACI, BAFF‑R can induce Ig class switching, whereas BCMA promotes survival of B type PCs.54 Dysregulation of BAFF production may be associated with development of autoimmune diseases.55 The role of the APRIL and BAFF proteins has been the subject of several publications, and is an important element in understanding the role of the BCMA receptor, the intracellular signal transduction cascade, and the potential drug resistance.56,57
G protein–coupled receptor class C group 5 member D
GPRC5D is located on chromosome 12p13. It is an orphan G protein‑coupled receptor, meaning its ligand is unknown, and, owing to the lack of in vivo models, its signaling mechanism and function in MM and other cells remains incompletely understood.58 GPRC5D is primarily expressed in PCs, hair follicles, skin, and the oropharynx. In contrast to CD38 or BCMA, GPRC5D is poorly expressed in T cells, NK cells, monocytes, granulocytes, bone marrow progenitors, and normal B cells but is present on PCs.59 The GPRC5D antigen seems to be a very good target for T‑cell redirection therapy, and the GPRC5D protein is also highly expressed in SMM patients.60 GPRC5D protein expression, in contrast to its mRNA expression, is restricted to epithelial structures of the skin, tongue, and CD138+ bone marrow cells studied in healthy donors.61 Among the targeted therapies available for MM patients, important aspects include the co‑occurrence of BCMA and GPRC5D receptors, the loss of one of them, the sequential use of treatment, and the reduction in receptor density. The expression patterns of BCMA and GPRC5D on CD138+ cells are independent of each other. Notably, the loss of BCMA observed in disease relapse is not associated with the loss of GPRC5D expression, making GPRC5D‑targeted therapy an interesting option for patients after treatment with BCMA‑targeted therapies.62 Owing to the availability of therapy and the multitude of clinical trials, the number of patients after BCMA therapy is constantly growing, and therapeutic options for this population are very limited. It seems that GPRC5D‑targeted drugs may be a solution for this group. In the literature, relapse of MM after GPRC5D‑targeted therapy was described in the case of biallelic GPRC5D mutation, loss of GPRC5D expression, and epigenetic silencing of promoter and enhancer regions.63
Overview of the most important clinical trials using cellular therapies: chimeric antigen receptor‑T cells
Idecabtagene vicleucel
Idecabtagene vicleucel (Ide‑cel, ABECMA) is a genetically modified autologous product based on T lymphocyte cells. Ex vivo transduction via a replication‑incompetent lentiviral vector (LVV) is used during the production process of modified T lymphocytes. The LVV encodes a CAR that recognizes the BCMA antigen, a single‑chain variable fragment directed against human BCMA. Additionally, the CAR receptor combines with the 4‑1BB costimulatory domain and the CD3-ζ signaling domain. The activation of T lymphocytes targeted to specific antigens by the Ide‑cel product results in the proliferation of T lymphocytes expressing the CAR, the secretion of cytokines, and, as a result, cytolytic activity, destroying cells expressing BCMA. The efficacy of Ide‑cel was assessed in a phase I study involving 33 patients with RRMM.64 Owing to promising results, the KarMMa‑2 phase II trial (An Efficacy and Safety Study of bb2121 in Subjects with Relapsed and Refractory Multiple Myeloma and in Subjects with High‑Risk Multiple Myeloma) included 128 patients (84% of whom were triple‑class refractory, and 26% were penta‑class refractory) and administered CAR‑T cells in variable amounts (150–450 × 10⁶).65 ORR was 73%, and the median PFS was 8.8 months in all treated patients.65 At the highest target dose (450 × 10⁶ CAR+ T cells), ORR increased to 81%, with a CR rate of 39%. The median PFS increased to 12.2 months with longer follow‑up, and the OS was 19.4 months. Among the patients who achieved CR or sCR, 79% also had MRD negative status at a sensitivity level of 1:10-5, and 59% of patients were MRD‑negative at 12 months. Typical complications of therapy include CRS, which occurs in 84% of patients, and neurotoxicity, which occurs in 18% of patients.65,66 In March 2021, the Food and Drug Administration (FDA) agency approved Ide‑cel for use in patients with RRMM who had received 4 or more prior LOT, owing to the breakthrough effectiveness of Ide‑cel. The phase III KarMMa‑3 trial (Efficacy and Safety Study of bb2121 Versus Standard Regimens in Subjects with Relapsed and Refractory Multiple Myeloma)67 compared Ide‑cel with standard treatment regimens in triple‑exposed (TCE) RRMM after 2–4 prior regimens (must have had previous IMiD, PI, and anti‑CD38 mAb). In the Ide‑cel group, 66% of the patients had triple‑class refractory disease, and 95% had daratumumab‑refractory disease. After median follow‑up of 30.9 months, the median PFS was significantly improved in the Ide‑cel cohort (13.8 months vs 4.4 months), with a 51% reduced risk of progression or death. Following these results, the FDA expanded the indication for Ide‑cel to TCE RRMM after at least 2 prior LOT.
Ciltacabtagene autoleucel
Ciltacabtagene autoleucel (Cilta‑cel, CARVYKTI) is a modified anti‑BCMA T‑cell therapy that contains 2 variable heavy‑chain domains in its CAR. These domains are highly specific for the BCMA antigen, which gives the receptor high avidity. One of the most important initial studies on the efficacy of Cilta‑cel was the CARTITUDE‑1 study68 (A Study of JNJ‑68284528, a Chimeric Antigen Receptor T Cell [CAR‑T] Therapy Directed Against B‑Cell Maturation Antigen [BCMA] in Participants with Relapsed or Refractory Multiple Myeloma). The study group consisted of 97 patients with RRMM (88% triple‑class refractory, 42% penta‑class refractory, with a median of 6 prior therapies), who received Cilta‑cel. ORR was 97%, and 67% of the patients achieved sCR. The 12‑month PFS rate was 77%, and the OS rate was 89%.68 CRS occurred in 95% of the patients (4% at the grade 3 or 4), while ICANS was observed in 21% (9% at the grade 3 or 4).68 In 2023, updated results were released after 33 months of follow‑up, showing a median PFS of 34.9 months and an estimated 62.9% OS at 36 months.69 In February 2021, the FDA approved Cilta‑cel for patients with RRMM who had received 4 or more prior LOT. The CARTITUDE‑2 study (A Study of JNJ‑68284528, a Chimeric Antigen Receptor T Cell [CAR‑T] Therapy Directed Against B‑cell Maturation Antigen [BCMA] in Participants with Multiple Myeloma) investigated the efficacy of CAR‑T cells in various MM patient populations.70,71 It is a multicenter, multicohort phase II study conducted in Europe and the USA. Cohort A included 20 MM patients with 1–3 prior LOT (median, 2). The cohort had 100% lenalidomide‑refractory patients, 35% individuals with high‑risk cytogenetics, and 40% of triple‑class refractory participants. As many as 85% of the patients had received prior auto‑HSCT. ORR was 95%, with 90% of patients achieving CR or better. After 24 months of follow‑up, the PFS and OS rates were both 75%. Typical complications included CRS (95%, with 10% at the grade 3 or 4), neutropenia (with 95% at the grade 3 or 4) anemia (75%, with 45% at the grade 3 or 4), and thrombocytopenia (80%, with 40% at the grade 3 or 4).70 Cohort B included 19 patients with early relapse (≤12 months after either ASCT or the start of initial treatment). The results in this cohort were impressive, with ORR of 100% and CR of 74%. At 24 months, the PFS rate was 73%, and the OS rate was 84%. Among the complications, CRS was reported in 84.2% of patients (grade 3 or 4 in only 1 patient), and neurotoxicity in 26.3% (grade 3 or 4 in only 1 patient).70 Cohort C included 20 patients with RRMM who had received prior treatment with noncellular BCMA‑targeted therapies (median LOT, 8). Results were somewhat less favorable, with ORR of 60%, and 55% of patients achieving at least very good partial response (VGPR). The median PFS was 9.1 months.71 Complications included CRS (60%, with no grade 3 or 4, ICANS (20%, with 10% grade 3 or 4), and significant hematologic toxicities. In the phase III CARTITUDE‑4 study,72 a CAR‑T therapy Cilta‑cel was compared with standard care regimens (pomalidomide, bortezomib, and dexamethasone [PVd] or daratumumab, pomalidomide, and dexamethasone [DPd]) in RRMM patients (100% lenalidomide‑refractory) after 1 to 3 prior LOT. The study included 419 patients (208 who received Cilta‑cel therapy and 211 who received standard care). Among the patients receiving Cilta‑cel therapy, 59.4% were high‑risk, and 14.4% were triple‑class refractory. Among those receiving standard care, 62.9% were high‑risk, with 15.6% triple‑class refractory.72 Only one‑fourth of patients had received prior CD38 antibody therapy. After median follow‑up of 15.9 months, ORR in the intent‑to‑treat group was 84.6% in the Cilta‑cel group vs 67.3% in the standard care group; ORR in the treated patients was 99%. The CR/sCR rate was 73.1% vs 21.8%, and the MRD‑negative rate was 60.6% vs 15.6%. The median PFS was not reached in the Cilta‑cel arm, while the PFS was 11.8 months in the standard care arm. CRS was observed in 76.1% of patients in the Cilta‑cel arm (1.1% at the grade 3 or 4), and ICANS was observed in 4.5% (0.1% at the grade 3 or 4).72 Table 2 provides a summary of the most important studies involving Ide‑cel and Cilta‑cel.
Trial | CAR‑T product | Phase | Patients, n | Patient population | Comparison | Key outcomes |
a The trial is planned to end in 2030; active, not recruiting.
b The reported results vary depending on the cohort analyzed; see text for details.
Abbreviations: CAR, chimeric antigen receptor; CR, complete remission; Depend on cohort: Number of patients depends on the patient group, more information in the main text; IMiD, immunomodulatory drug; MM, multiple myeloma; NA, not available; NDMM, newly‑diagnosed multiple myeloma; ORR, overall response rate; OS, overall survival; PFS, progression‑free survival; PI, proteasome inhibitor; RRMM, refractory / relapse multiple myeloma; SoC, standard of care; T, T cell | ||||||
KarMMa65 | Abecma (Ide‑cel) | 2 | 128 | RRMM after ≥3 prior therapies, including PI, IMiD, anti‑CD38 | Single‑arm | ORR 73%, CR 33%, PFS 8.8 months, OS 19.4 months |
KarMMa‑2a | Abecma (Ide‑cel) | 2 | Enrollment (estimated) 264, depending on cohort | Early RRMM after 1–2 prior lines, post‑transplant relapse | Single‑arm | Evaluating safety and efficacy in earlier‑line use |
KarMMa‑367 | Abecma (Ide‑cel) | 3 | 254 (Ide‑cel), 132 SoC | RRMM after 2–4 prior lines, including IMiD, PI, anti‑CD38 | Ide‑cel vs SoC | PFS 13.3 months (vs 4.4 months SoC), ORR 71% (vs 42%) |
CARTITUDE‑169 | Carvykti (Cilta‑cel) | 1b/2 | 97 | RRMM after ≥3 prior therapies, including IMiD, PI, anti‑CD38 | Single‑arm | ORR 98%, mPFS 34.9 months, mOS: not reached (36‑month rate: 62.9%) |
CARTITUDE‑270,71 | Carvykti (Cilta‑cel) | 2 | Depending on cohort | RRMM with 1–3 prior lines, including post‑transplant relapse | Single‑arm | High efficacy in less pretreated patientsb |
CARTITUDE‑472 | Carvykti (Cilta‑cel) | 3 | 419
(208 Cilta‑cel and 211 SoC) | 1–3 prior therapies, including lenalidomide‑resistant MM | Cilta‑cel vs SoC | The 74% reduction in risk of progression or death, along with the high rates of CR and MRD negativity, underscore Cilta‑cel’s strong potential to become a pivotal treatment option for patients with MM following their first relapse |
Anitocabtagene autoleucel
Anitocabtagene autoleucel (Anito‑cel/CART‑ddBCMA) is an autologous T‑cell product that is genetically modified ex vivo to express a D‑domain chimeric CAR, followed by a CD8 hinge and transmembrane region, which is fused to intracellular signaling domains of 4‑1BB and CD3ζ. Anito‑cel contains CAR+ CD3+ T cells, which undergo T‑cell activation, gene transfer using a replication‑deficient LVV, and subsequent expansion. In the iMMagine‑1 study73 (Study of Anitocabtagene‑autoleucel in Relapsed or Refractory Multiple Myeloma), a phase 2 pivotal trial, patients with RRMM who had received at least 3 prior LOT were enrolled and received a single infusion of Anito‑cel. Preliminary results from 86 patients showed a 97% ORR, with 62% of patients achieving CR or sCR at median follow‑up of 9.5 months. The median age of participants was 66 years, with a median of 4 prior LOT. Notably, 45% of patients had received 3 prior LOT, 69% were classified as triple‑class refractory, and 34% were penta‑class refractory. Among the patients evaluable for MRD, 92% (36 out of 39) achieved MRD negativity at a sensitivity level of at least 10-5. The 6‑month PFS rate was 90%, and OS rate was 95%, though the median PFS and OS have not yet been reached.73 CRS was observed in 49 patients (84%), with 79% experiencing either no CRS (16%) or grade 1 CRS (64%). Only 19% of patients had grade 2 CRS. Importantly, no delayed or non‑ICANS neurotoxicity, such as parkinsonism, cranial nerve palsies, or Guillain–Barré syndrome have been associated with Anito‑cel treatment to date. This study demonstrated that Anito‑cel provides deep and durable responses with a predictable and manageable safety profile, particularly in high‑risk RRMM populations, including those with triple- and penta‑class refractory disease.
Teclistamab
Teclistamab was the first bispecific antibody approved for the treatment of RRMM in patients who had received at least 4 prior LOT, including a PI, an IMiD, and an anti‑CD38 mAb. The drug’s approval was based on the landmark MajesTEC‑1 study (A Study of Teclistamab in Participants with Relapsed or Refractory Multiple Myeloma), published in 2022.74 In this study, 165 patients received teclistamab (median LOT, 5), with 77.6% being triple‑class refractory and 26% having high‑risk cytogenetics. ORR was 63%, with 65 patients (39.4%) achieving a CR or better. Reported adverse events included CRS in 72.1% of patients (grade 3, 0.6%; no grade 4 cases), ICANS was observed in 5 patients (3%), all of whom had grade 1 or 2 events. A clinically significant complication of teclistamab use was infection, which occurred in 76.4% of the patients (grade 3 or 4, 44.8%). Regarding long‑term outcomes, after 22 months of follow‑up, 43% of the patients had achieved CR or sCR, the medina PFS (mPFS) was 12.5 months, while the median OS (mOS) was 21.9 months and median duration of response (mDOR) was 24 months.75 In the phase 1/2 MajesTEC‑1 study, a cohort of 40 patients with prior exposure to BCMA‑targeted therapies, including ADC or CAR‑T‑cell therapy (ADC, n = 29; CAR‑T, n = 15; both, n = 4), was enrolled to evaluate the efficacy of teclistamab in individuals previously treated with anti‑BCMA agents. At median follow‑up of 28 months, ORR was 52.5%, 47.5% of the patients achieved at least VGPR, and 30% at least CR. The mDOR was 14.8 months, mPFS 4.5 months, and mOS 15.5 months.76
Elranatamab
Elranatamab is a humanized bispecific IgG2 anti‑BCMA/CD3 antibody that was approved by the FDA in August 2023 for the treatment of patients with RRMM who had received at least 4 prior LOT, including a PI, an IMiD, and an anti‑CD38 mAb. The FDA approval was based on the efficacy demonstrated in the phase 2 MagnetisMM‑3 study (Study Of Elranatamab [PF‑06863135] Monotherapy in Participants With Multiple Myeloma Who Are Refractory to at Least One PI, One IMiD, and One Anti‑CD38 mAb).77 In this study, elranatamab was administered at escalating doses to a target dose of 76 mg. Among the 123 patients who received elranatamab, 96.7% had received at least 3 prior LOT (median, 5) and 42.3% were penta‑refractory.78 Regarding long‑term efficacy and safety, 61% of patients achieved ORR, while 35.8% achieved at least CR. Among the patients with at least CR, 89.7% were MRD negative. The median DOR, PFS, and OS were not reached in the study. A retrospective analysis showed that at 15 months, the probability of maintaining a response, remaining progression‑free, and surviving were 70.8%, 50.2%, and 56.3%, respectively.78,79 Elranatamab had an acceptable safety profile. The most common adverse events included infections in 69.9% of patients (grades 3 or 4, 40.7%), CRS in 57.7% (grade 3 or 4, 0%), peripheral neuropathy in 17.1%, or ICANS in 3.4% (grade 3 or 4, 0%).77 In an analysis of patients with RRMM who had prior exposure to BCMA‑directed therapies, elranatamab demonstrated both efficacy and a manageable safety profile. Among the patients previously treated with BCMA‑targeted therapy, elranatamab emerged as a viable therapeutic option. This cohort represents a particularly challenging population with limited treatment alternatives, making the observed efficacy of elranatamab a significant and encouraging finding. In a pooled analysis from the MagnetisMM studies, among patients who had received BCMA‑directed ADC (67.4%), CAR T‑cell therapy (41.9%), and both modalities (9.3%), the median follow‑up was 10.3 months. The median duration of treatment with elranatamab was 3.3 months. ORR was 45.3%, with CR or better achieved in 17.4% of the patients. DOR at 9 months was 67.3% for the patients previously treated with BCMA‑directed ADCs, and 78.9% for those who had received prior CAR T‑cell therapy. ORR in these subgroups was 41.4% and 52.8%, mPFS was 4.8 months, while mOS had not been reached at 10 months, with an estimated 9‑month OS rate of 60.1%.80
Talquetamab
Talquetamab is the first bispecific IgG4 antibody targeting the GPCR5D receptor on myeloma cells and the CD3 receptor on T lymphocytes.81 It was evaluated in the MonumenTAL‑1 study82 (A Study of Talquetamab in Participants with Relapsed or Refractory Multiple Myeloma), which included patients who had received at least 4 prior LOT. Talquetamab was administered as monotherapy with 2 dosing regimens: 0.4 mg/kg once weekly (QW) and 0.8 mg/kg once every 2 weeks (Q2W). ORR was 74% in the QW group and 73% in the Q2W group. In the QW group, 58% patients achieved at least VGPR and in the Q2W group, 57% patients achieved at least VGPR.82 Talquetamab demonstrated durable responses. In the 0.8 mg/kg Q2W cohort, the mDOR was 17.5 months while in the 0.4 mg/kg QW cohort, the mDOR was 9.5 months.83,84 The FDA approved talquetamab in August 2023 based on data from the MonumenTAL‑1 study.85 CRS occurred in 77% patients (grade 3 or 4, 3%) in the QW group and 80% (grade 3 or 4, 0%) in the Q2W group. Infections occurred in 47% of the patients in the QW group (grade 3 or 4 infections in 2 out of 30 patients) and in 34% in the Q2W group (grade 3 or 4 infections in 3 out of 44 patients). Treatment‑related neurotoxic events occurred in 10% of the patients in the QW group and in 5% in the Q2W group.85 The MonumenTAL‑1 study included a cohort of 78 patients with prior exposure to BsAb or CAR T‑cell therapy, of whom 94% received prior BCMA‑directed treatment. In this heavily pretreated patients group, at median follow‑up of 20.5 months, ORR was 66.7%, with at least CR in 42.3%.83,84 The TRIMM‑2 study86 (A Study of Subcutaneous Daratumumab Regimens in Combination with Bispecific T Cell Redirection Antibodies for the Treatment of Participants with Multiple Myeloma) investigated the efficacy of combining talquetamab with daratumumab in RRMM patients. The study included patients who had received at least 3 LOT, had been previously treated with PI and IMiD, or were refractory to PI and IMiD but had not received anti‑CD38 therapy within up to 90 days prior to study entry. Among patients not previously treated with anti‑CD38 therapy, ORR was 78% (66% ≥VGPR; 45% ≥CR). The study also showed that prior exposure to BsAb did not significantly affect response rates, with ORR of 75% regardless of prior BsAb use. Among CAR T‑cell–exposed and CAR T‑cell–refractory patients, ORR was 74% and 64%, respectively. TRIMM‑2 demonstrated promising efficacy and an acceptable safety profile.86
Linvoseltamab
Linvoseltamab is a BCMA × CD3 BsAb. The efficacy of this drug was evaluated in a study involving 117 patients (39% high‑risk MM and 28% penta‑class refractory).87 This phase 1/2 study assessed the efficacy and safety of 50 mg and 200 mg doses. At median follow‑up of 14.3 months, ORR was 71%, with 50% of patients achieving at least CR. In the 104 patients treated with 50 mg dose, ORR at median follow‑up of 7.4 months was 48%, with 21% of the participants achieving at least CR. The mDOR for the 200 mg cohort was 29.4 months. Linvoseltamab represents another effective treatment option for RRMM.87
Cevostamab
Cevostamab is an Fc receptor‑homolog 5 (FcRH5) × CD3 BsAb. The efficacy of cevostamab was assessed in a cohort of 167 patients (38% high‑risk MM, 28% with extramedullary disease, median LOT of 6, 95.8% triple‑class refractory, 73.7% penta‑drug refractory, 57% after at least 1 prior BCMA‑targeted therapy). With median follow‑up of 11.3 months, ORR was 43.1%, with 6.6% patients achieving sCR, 12.6% VGPR, and 17.4% PR.88 The VGPR or better rate was 25.7%, and the mDORwas 10.4 months. Among the patients who had received at least 1 prior BCMA‑targeted therapy, ORR was 30.2%, while in those without prior BCMA‑targeted therapy, ORR was 60.6%. CRS occurred in 74.3% of the patients (grade 3 or 4, 1.8%), and ICANS was observed in 13.2% (grade 3 or 4, 1.8%). Cevostamab is a therapeutic option for patients with heavily pretreated RRMM.88
Side effects
The use of immunotherapies can be associated with potentially severe and life‑threatening complications, including CRS, ICANS, delayed neurotoxicity, cytopenias, and infection.
Cytokine release syndrome
CRS develops due to T‑cell activation, proliferation, and inflammation and is a common complication of CAR‑T cell therapy and TCE. Clinical symptoms include fever, hypoxia, and hypotension, which, in severe cases, can progress to shock and multiorgan failure.89 Laboratory abnormalities typically include elevated C‑reactive protein, ferritin, and lactate dehydrogenase levels, along with coagulation disorders. The severity of CRS is classified based on the American Society of Transplantation and Cellular Therapy grading system.90 Approximately 60% of patients receiving TCE and most patients receiving CAR T‑cell therapy develop CRS, although CRS of grade 3 or higher is less common.90 The management of CRS depends on its severity. For example, to mitigate the risk of CRS when initiating teclistamab therapy, hospitalization for 48 hours after each dose escalation and the first full dose is recommended. Management of CRS is outlined in Table 3. The most commonly used drugs for treating CRS are tocilizumab and corticosteroids.91,92 CRS that is refractory to tocilizumab and steroids remains a clinical challenge. In the CARTITUDE‑1 study,69 the use of anakinra, anti‑interleukin‑1 (IL‑1) monoclonal antibody (administered every 8–12 h for 2.5 days) successfully resolved CRS in 17 of 18 patients. Potential alternative treatments for refractory CRS include, for example, siltuximab (anti‑IL‑6 monoclonal antibody).93 Research is ongoing to optimize CRS prevention strategies, including prophylactic administration of tocilizumab or dexamethasone.94,95
CRS grade | Signs and manifestations | Treatment |
Abbreviations: BiPAP, bilevel positive airway pressure; CPAP, continuous positive airway pressure; CRS, cytokine release syndrome; IV, intravenous | ||
Grade 1 | Temperature ≥38 °C Hypotension: absent
Hypoxia: absent |
|
Grade 2 | Temperature ≥38 °C
Hypotension: not requiring vasopressors
Hypoxia: requiring low‑flow nasal cannula (oxygen ≤ 6 l/min) or blow‑by |
|
Grade 3 | Temperature ≥38 °C
Hypotension: requiring one vasopressor with or without vasopressin and / or
Hypoxia: requiring high‑flow nasal cannula (oxygen > 6 l/min), facemask, nonrebreather mask or Venturi mask |
|
Grade 4 | Temperature ≥38 °C
Hypotension: requiring multiple vasopressors (excluding vasopressin)
Hypoxia: requiring positive pressure (eg, CPAP, BiPAP, intubation and mechanical ventilation) |
|
Immune effector cell‑associated neurotoxicity syndrome and delayed neurotoxicity
Neurotoxicity is another significant complication of anti‑MM therapies, often manifested as ICANS. This condition typically arises within the first few days following BsAb or CAR‑T cell therapy, but delayed neurotoxicity has also been observed. Common symptoms of ICANS include headache, confusion, and disorientation, dysgraphia, difficulty concentrating, and expressive aphasia, apraxia, altered consciousness, seizures, and muscle weakness. The median onset of ICANS is 2 days after Ide‑cel infusion and 8 days after Cilta‑cel infusion. Neuroimaging findings are usually normal but some cases may show cerebral edema or hyperintensities within the limbic system and brainstem. Delayed neurotoxicity has been reported, particularly with Cilta‑cel, including neuropathy, cranial nerve palsy (most commonly affecting the facial nerve), and most concerning a frequently irreversible Parkinsonism‑like syndrome (movement and neurocognitive treatment‑emergent adverse events [MNTs]). A real‑world study of Cilta‑cel reported 10% delayed neurotoxicity, with cranial nerve palsy being the most common symptom.96 In a retrospective analysis published in 2025, encompassing data from 19 leading medical institutions, no significant difference in the incidence of ICANS was observed between Ide‑cel and Cilta‑cel.97 The exact mechanism of neurotoxicity remains unclear. However, interactions between CAR‑T cells and myeloid cells are believed to drive proinflammatory cytokine production, leading to disruption of the blood‑brain barrier and endotheliopathy. Another concern is that BCMA is expressed in the central nervous system (CNS), particularly in the pons and midbrain. This, together with increased permeability, allows cytokines to enter the CNS, resulting in inflammation and neurotoxicity. Studies are currently investigating the underlying factors contributing to ICANS. Risk factors for development of ICANS induce large expansion of CAR‑T cells, high disease burden, and male sex (observed in CARTITUDE‑1 and CARTITUDE‑4, where remarkably all MNT cases occurred in men).98 Management of ICANS is detailed in Table 4. Comparison of the toxicity profiles of Ide‑cel vs Cilta‑cel is presented in Table 5, and Table 6 shows the toxicity profiles of BsAbs.
Abbreviations: EEG, electroencephalogram; ICANS, immune effector cell‑associated neurotoxicity syndrome; ICE, immune effector cell‐associated encephalopathy | |
ICANS | Symptoms and manifestation |
Grade 1 | ICE score 7–9
Fatigue, awakens spontaneously
Seizure: absent |
Grade 2 | ICE score 3–6
Awakens to voice
Seizure: absent |
Grade 3 | ICE score 0–2
Awakens only to tactile stimulus
Seizure: any clinical seizure focal or generalized that resolves rapidly, or nonconvulsive seizures on EEG that resolve with intervention
Cerebral edema: focal / local edema on neuroimaging (without bleeding) |
Grade 4 | Patient unable to perform ICE score
Patient unarousable or requires vigorous stimuli
Seizure: life‑threatening prolonged seizure (>5 min) or repetitive clinical or electrical seizures without return to baseline in between
Deep focal motor weakness such as hemiparesis or paraparesis
Diffuse cerebral edema on neuroimaging; decerebrate or decorticate posturing; or cranial nerve VI palsy or papilledema or Cushing’s triad |
ICE score | |
Orientation | Orientation to year, month, city, hospital: 4 points |
Naming | Ability to name 3 objects (eg, window, pen, stethoscope): 3 points |
Following commands | Ability to follow simple commands (eg, Show me 2 fingers): 1 point |
Writing | Ability to write a standard sentence (eg, Home sweet home): 1 point |
Attention | Ability to count backwards from 100 by 10: 1 point |
Parameter | Cilta‑Cel (Ciltacabtagene Autoleucel) | Ide‑Cel (Idecabtagene Vicleucel) |
Overall ICANS incidence | About 20% | About 25% |
Grade ≥3 ICANS incidence | About 2% | About 2% |
Median time to onset ICANS, d | 8 (range, 1–28) | 3 (range, 1–12) |
Parkinsonism incidence | About 3% | No parkinsonism reported in the KarMMa trials65 |
Overall CRS incidence | About 81% | About 85% |
None | About 19% | About 15% |
Grade 1 | About 48% | About 56% |
Grade 2 | About 24% | About 25% |
Grade 3 | About 5% | About 4% |
Grade 4 | About 5% | <1% |
Median time to onset CRS, d | About 3 (range, 1–15) | About 1 (range, 1–18) |
Tocilizumab utilization | About 71% | About 67% |
Median duration, d | 4 (range, 1–97) | 5 (range, 1–63) |
Hypogammaglobulinemia | Hypogammaglobulinemia (IgG <0.5 g/l) across CARTITUDE‑1 and CARTITUDE‑4: 94%69,72 | Hypogammaglobulinemia (IgG <0.5 g/l) across KarMMa and KarMMa‑3: 45%65,67 |
Hemophagocytic lymphohistiocytosis / macrophage activation syndrome
Hemophagocytic lymphohistiocytosis / macrophage activation syndrome (HLH/MAS) is a rare complication of CAR‑T therapy, characterized by excessive activation of cytotoxic T cells, NK cells, and macrophages. This hyperactivation leads to massive cytokine production, which can result in multiorgan failure.99 HLH/MAS develops as cytokine storm, with proinflammatory cytokines likely including IL‑1, IL‑6, IL‑12, IL‑18, IL‑27, MIG/CXCL9, IP10/CXCL10, I‑TAC/CXCL11, MCP‑1/CCL2, MIP‑1α/CCL3, GM‑CSF, interferon-γ, TNF-α, and IL‑8/CXCL8.100 This rare but much feared complication is associated with high mortality, and distinguishing between severe CRS and CAR‑T–related HLH/MAS can be challenging. Guidelines are available that propose diagnostic criteria for differentiation.99 In therapeutic management, most experts recommend anti–IL‑6 therapy and corticosteroids, with consideration for the addition of etoposide if no clinical improvement is observed after 48 hours.100 Etoposide acts by eliminating activated T lymphocytes, thereby reducing the production of proinflammatory cytokines.
Secondary primary malignancies
Secondary primary malignancies (SPMs) have been observed following CAR‑T therapy. In a recent study,101 malignancies were reported as adverse events in 536 of 12 394 patients (4.3%) with myelodysplastic syndromes and skin cancers, being the most commonly observed SPMs. However, precise data on SPMs after targeted immune therapies in MM remain limited.
Conclusions and future directions
In recent years, enormous progress has been made in the treatment of MM. However, significant disparities remain in the availability of new therapies across different regions and health care jurisdictions. Government and health care experts should work to reduce these gaps in access, primarily by expanding modern hospital networks and increasing the number of departments offering cell therapies as well as managing costs. Despite the promising efficacy of the therapies described above, they are not without serious and potentially life‑threatening side effects. Moreover, high price of the treatment and limited number of hospitals providing molecular‑targeted and immune therapies make rapid improvements in access unlikely in many countries. An emerging challenge in clinical practice is the increasing number of patients who become resistant to novel molecular‑targeted therapies but, fortunately, the therapeutic armamentarium continues to expand, both with immune therapy and other novel agents including small molecules. As this number continues to grow in the coming years, it underscores the urgent need to enhance existing treatments, optimize supportive care, minimize complications and refine treatment sequencing for newly‑diagnosed and relapsed disease, with strategic considerations paramount for individual patients, thus providing truly tailored care. Further research and innovation will be essential to improve patient outcomes in MM but continued progress and achievement of functional cure in an increasing proportion of patients appears now more possible than ever before.
- Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014; 15: e538‑e548. | Crossref
- Cowan AJ, Allen C, Barac A, et al. Global burden of multiple myeloma: a systematic analysis for the global burden of disease study 2016. JAMA Oncol. 2018; 4: 1221‑1227. | Crossref
- Mohty M, Facon T, Malard F, et al. A roadmap towards improving outcomes in multiple myeloma. Blood Cancer J. 2024; 14: 135. | Crossref
- Zhu DT, Park A, Lai A, et al. Multiple myeloma incidence and mortality trends in the United States, 1999‑2020. Sci Rep. 2024; 14: 14564. | Crossref
- Bergstrom D, Kotb R, Louzada ML, et al. Consensus Guidelines on the diagnosis of multiple myeloma and related disorders: recommendations of the Myeloma Canada Research Network Consensus Guideline Consortium. Clin Lymphoma Myeloma Leuk. 2020; 20: 352‑367. | Crossref
ARTICLE INFORMATION