Neuroendocrine carcinoma of the extrahepatic bile duct in a patient with primary sclerosing cholangitis: a rare and unexpected diagnosis

 CC BY 4.0
CC BY 4.0
Neuroendocrine carcinoma of the extrahepatic bile duct in a patient with primary sclerosing cholangitis: a rare and unexpected diagnosis
A 30‑year‑old woman with a medical history significant for primary sclerosing cholangitis (PSC) and ulcerative colitis was referred to a hepatology department with suspected ampullary carcinoma. One year earlier, she had undergone radical resection of sigmoid adenocarcinoma, and the next year, she had 2 episodes of cholangitis, necessitating endoscopic stenting.
Upon admission, the patient was in a good general condition and asymptomatic. Biochemical evaluation showed evidence of biliary duct injury: alkaline phosphatase of 201 IU/l (reference range [RR] <120 IU/l) and γ-glutamyl transferase of 121 IU/l (RR <50 IU/l). However, aminotransferases, total bilirubin, carcinoembryonic antigen, and carbohydrate antigen 19–9 levels were all within normal limits.
Magnetic resonance cholangiopancreatography showed significant common bile duct stenosis (Figure 1A) and enlarged lymph nodes within the hepatic hilum. The patient subsequently underwent endoscopic retrograde cholangiopancreatography with double stenting and tissue sampling. Histopathological examination identified poorly differentiated malignant cells, raising concern for cholangiocarcinoma. Positron emission tomography excluded recurrence of colorectal carcinoma and did not show any other suspicious intra‑abdominal lesions.
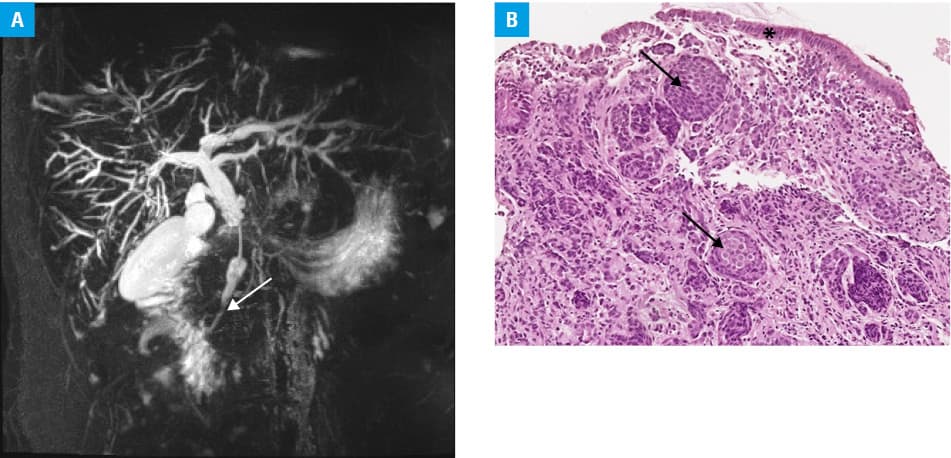
With a presumptive diagnosis of cholangiocarcinoma, the patient underwent pancreatoduodenectomy using the Traverso–Longmire technique. Unexpectedly, the postoperative histopathological assessment identified neuroendocrine carcinoma (NEC) of the distal extrahepatic bile duct (Figure 1B), with no regional lymph node involvement, but a positive radial resection margin (classified as pT2N0R1 according to the American Joint Committee on Cancer, 8th edition). The patient received 6 cycles of adjuvant etoposide and cisplatin chemotherapy. Follow‑up imaging showed no evidence of recurrent disease. Eight months later, disease progression was detected with a new para‑aortic lymph node lesion (30 mm × 22 mm) and a left adrenal metastasis (16 mm × 13 mm). Second line folinic acid, fluorouracil, and irinotecan chemotherapy with radiotherapy, targeting retroperitoneal lymph nodes, was initiated. Post‑treatment imaging demonstrated a partial radiologic response, and the patient remains clinically stable to date, 3 years postdiagnosis.
Gastroenteropancreatic neuroendocrine neoplasms (NENs) are classified as well‑differentiated neuroendocrine tumors and poorly differentiated NECs.1 Primary NENs of the liver and extrahepatic biliary tree are exceptionally rare and often present diagnostic challenges due to nonspecific symptoms, such as right upper quadrant pain, jaundice, or a palpable abdominal mass.2
Two cases describing an association between NENs and PSC have been previously reported: one involving a hepatic composite tumor exhibiting both neuroendocrine and cholangiocarcinomatous differentiation, and another documenting a primary hepatic NEN.3,4 However, to the best of our knowledge, this is the first reported case of NEC arising in the context of PSC.
NECs of the biliary tract are frequently accompanied by non‑neuroendocrine components. Recent molecular studies on gallbladder NECs have demonstrated frequent RB1 loss, p16 overexpression, and, in some instances, TP53 mutations—molecular alterations commonly observed in autochthonous adenocarcinomas.5 When considered alongside the well‑established risk of developing cholangiocarcinoma in PSC patients, this molecular convergence raises the possibility of a shared tumorigenic pathway within this pathological setting.
This observation underscores the need for further molecular and clinicopathological studies to elucidate the potential interplay between chronic biliary inflammation and neuroendocrine tumorigenesis in patients with PSC.
- Rindi G, Mete O, Uccella S, et al. Overview of the 2022 WHO Classification of Neuroendocrine Neoplasms. Endocr Pathol. 2022; 33: 115‑154. | Crossref
- Modlin IM, Shapiro MD, Kidd M. An analysis of rare carcinoid tumors: clarifying these clinical conundrums. World J Surg. 2005; 29: 92‑101. | Crossref
- Koplin S, Agni R. Hepatic composite tumor in a patient with primary sclerosing cholangitis. Pathol Res Pract. 2009; 205: 361‑364. | Crossref
- Bel Haj Yahia D, Bel Hadj Yahya M, Chelly I, et al. 21 years of evolution of primary hepatic neuroendocrine neoplasm in a patient with primary sclerosing cholangitis: a case report. Int J Surg Case Rep. 2023; 106: 108205. | Crossref
- Luchini C, Pelosi G, Scarpa A, et al. Neuroendocrine neoplasms of the biliary tree, liver and pancreas: a pathological approach. Pathologica. 2021; 113: 28‑38. | Crossref
ARTICLE INFORMATION