Resistant thrombotic thrombocytopenic purpura successfully treated with caplacizumab in a patient with acute myeloid leukemia



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Resistant thrombotic thrombocytopenic purpura successfully treated with caplacizumab in a patient with acute myeloid leukemia
Thrombotic thrombocytopenic purpura (TTP) is a rare blood disorder presenting with microangiopathic hemolytic anemia, thrombocytopenia, and a reduction in disintegrin and metalloproteinase with a thrombospondin type‑1 motif, member 13 (ADAMTS13) activity leading to the formation and accumulation of ultralarge von Willebrand factor (vWF) multimers and, as a result, excessive platelet aggregation. Numerous chemotherapeutics have been shown to induce thrombotic microangiopathies; thus, there have been reports of patients with TTP and stomach, breast, or lung cancer.1 The initial treatment includes steroids, therapeutic plasma exchange, rituximab, and caplacizumab—a vWF inhibitor.2
We present a case of a 42‑year‑old man, previously diagnosed with TTP, who was admitted to our ward for further treatment after plasmapheresis and steroid therapy failure. Before the diagnosis, the patient had been experiencing numerous neurological symptoms, including confusion and stroke‑like symptoms despite an unremarkable computed tomography (CT) scan. Physical examination showed tachycardia, skin paleness, intensive purpura, hematomas, and petechiae. Laboratory findings indicated microangiopathic hemolytic anemia with negative direct antiglobulin tests, thrombocytopenia, reticulocytosis, as well as elevated levels of lactate dehydrogenase and indirect bilirubin (Figure 1A–1C). ADAMTS13 activity was undetectable, with ADAMTS13 inhibitor levels of 9.1 IU/ml (reference range <0.5 IU/ml). Additionally, acute renal failure and respiratory distress were diagnosed. On admission, the patient received steroids and plasmapheresis combined with rituximab, with no improvement. Also, exacerbation of neurological and psychiatric symptoms, such as aggressiveness not responsive to sedatives, was observed. A repeated CT scan of the head showed no abnormalities. Subsequently, caplacizumab was administered, with gradual clinical improvement. Laboratory findings showed increased platelet count and ADAMTS‑13 activity (98%), and a decrease in reticulocytosis. Due to progressive neutropenia (Figure 1A), bone marrow biopsy was performed (Figure 1D and 1E), leading to an unexpected diagnosis of acute myeloid leukemia (AML) with maturation (according to the 2022 World Health Organization classification), with normal karyotype and no molecular abnormalities (intermediate risk according to the European Leukemia Network recommendations3). Complete remission (CR) with positive measurable residual disease (MRD) was achieved after standard induction therapy. In postremission therapy, consolidation with cytarabine and bone marrow transplant from a matched unrelated donor was performed. At the last follow‑up appointment (March 2025; 7 weeks after transplant), the patient remained in CR MRD(–) and had no TTP symptoms.
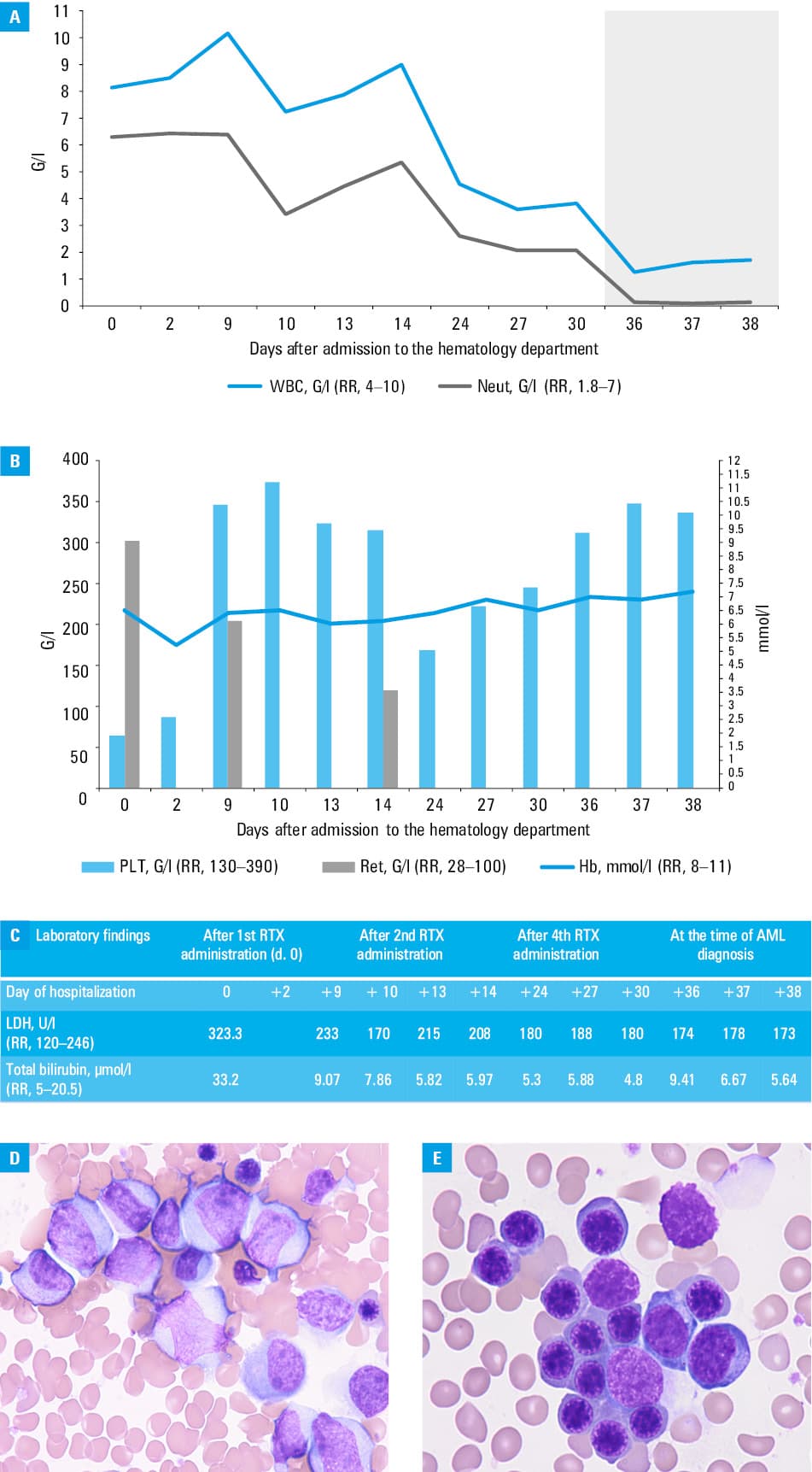
Coexistence of TTP and AML is extremely rare, and only a few cases have been reported worldwide.4,5 To our knowledge, this is the first such case in Poland. Low ADAMTS13 activity and the presence of ADAMTS13 antibodies, along with clinical presentation, suggested that TTP preceded AML. Adimora and Rojas Hernandez5 reported a similar patient; however, in that case, TTP was diagnosed following the confirmed AML. Initially, there was no suspicion of another hematological disease given the symptoms and laboratory results typical of TTP. In challenging cases, bone marrow biopsy remains the cornerstone of diagnostic strategy, and our case supports its key significance.
- Valério P, Barreto JP, Ferreira H, et al. Thrombotic microangiopathy in oncology – a review. Transl Oncol. 2021; 14: 101081. | Crossref
- Kurek M, Pasieka P, Włudarczyk A, Szczeklik W. When malaria imitates thrombotic thrombocytopenic purpura: importance of patient history and physician awareness of uncommon diseases. Pol Arch Intern Med. 2023; 133: 16569. | Crossref
- Mrózek K. Molecular cytogenetics in acute myeloid leukemia in adult patients: practical implications. Pol Arch Intern Med. 2022; 132: 16300. | Crossref
- Kucharik MP, Waldburg D, Chandran A, et al. Acute myeloid leukemia presenting as thrombotic thrombocytopenic purpura. Cureus. 2020; 12: e6869. | Crossref
- Adimora I, Rojas Hernandez CM. Thrombotic thrombocytopenic purpura triggered by acute myeloid leukemia: a case report. Am J Case Rep. 2022; 23: e935911. | Crossref
ARTICLE INFORMATION